
Abstract
Achondrogenesis type II (ACG2) is a lethal skeletal disorder caused by pathogenic variants in COL2A1. We present a fetus with cystic hygroma and severe shortening of the limbs at 14 weeks of gestation. We performed postnatal genetic analysis of the parents and fetus to diagnose the disease. A novel missense variant of COL2A1 [NM_001844.5: c.2987G>A, (p. Gly996Asp)] was identified, which led to the ACG2 diagnosis.
Similar content being viewed by others

COL2A1 is located on chromosome 12q13.11, encodes the alpha 1 chain of procollagen type 2, and is expressed primarily in cartilage1. Abnormalities in COL2A1 can cause several heritable diseases, such as achondrogenesis type II (ACG2) (OMIM #200610), hypochondrogenesis, platyspondylic dysplasia (the Torrance type), spondyloepiphyseal dysplasia congenita (SEDC), spondyloepimetaphyseal dysplasia (SEMD) (Strudwick type), Kniest dysplasia, Stickler syndrome, and spondyloperipheral dysplasia1. ACG2 is the most severe disease, and affected fetuses are stillborn or die immediately after birth.
There are approximately 461 skeletal disorders, of which more than 100 are identified in utero2,3. They are difficult to diagnose and show a low prenatal diagnostic rate (approximately 30–70%) in fetal ultrasonography3. In this study, we report a case of cystic hygroma and shortening of the limbs in a fetus. A novel heterozygous missense variant of COL2A1 was identified in the fetus, confirming the ACG2 diagnosis.
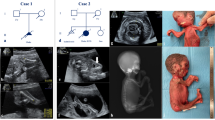
A Japanese couple was referred to our hospital for a thorough examination at 14 weeks and 2 days of gestation after being diagnosed with a cystic hygroma in the fetus at 12 weeks. They were a non-consanguineous healthy couple with a 30-year-old female and a 29-year-old-male. They had no family history of skeletal disorder (Fig. 1). It was their first spontaneous conception. No deformation of the cranial bone due to ultrasound probe compression was observed. The thickness of the cystic hygroma was 10.9 mm (Fig. 2a). Thoracic hypoplasia was suggested. The femur and humerus lengths, which were 4.5 mm (−4.1 SD) and 5.0 mm (−4.2 SD), respectively, revealed marked shortening of the limbs (Fig. 2b). The fetus was considered to have a severe skeletal disorder, but a detailed diagnosis could not be made at the time. Chromosomal analysis by amniocentesis and another fetal ultrasonography was suggested, but the couple insisted on terminating the pregnancy. At 14 weeks and 6 days of gestation, an induced abortion was performed. The infant weighed 40 g, was 10.5 cm long, and had marked limb shortening and a cystic hygroma (Fig. 2c, d).

A family pedigree of the Japanese family affected with achondrogenesis type II. P: proband.

a Ultrasonography of the fetus at 14 weeks of gestation showing a cystic hygroma of 10.9 mm and b a femur length of 4.5 mm (−4.1 SD), depicting marked shortening. c, d Clinical image of the affected fetus. The fetus had markedly shortened limbs and a cystic hygroma. e DNA sequences of II-3, II-6, and III-1 to detect the COL2A1 variant. The arrow indicates the novel variant that we detected in III-1.
Radiography taken after delivery showed marked shortening of the extremities. However, a detailed examination of ossification was difficult due to the fetus’s immaturity. Chromosomal analysis (G-banding) of the products of conception revealed a normal female karyotype (46,XX).
Following genetic counseling of the couple and obtaining written consent, we performed whole-exome sequencing (WES) for a disease diagnosis. The study was approved by the Institutional Review Board (IRB) of the National Center for Child Health and Development and the Jikei University School of Medicine [IRB number: 234 and IRB number: 27-060 (7945)].
DNA was extracted from the peripheral blood samples of the couple (designated II-3 and II-6 as per pedigree chart: Fig. 1) and the umbilical cord of the infant (III-1) using a previously described method4. A whole-exome library was prepared from the DNA samples of III-1 using the Agilent SureSelect v6 Capture Kit (Agilent Technologies, Santa Clara, CA, USA) following the manufacturer’s protocol. The libraries were sequenced on a HiSeq2500 (Illumina, San Diego, CA, USA) in the 101 bp paired-end mode. Sequence reads were mapped and aligned to the reference genome sequence hs37d5. Multisample calling of single nucleotide variations and short indels was performed with the RefSeq gene database in combination with 12 in-house control datasets. We extracted 1020 variants using our previously described method4. In addition, we extracted the variants with minor allele frequency (<0.01) according to the Integrated Japanese Genome Variation Database (ToMMo (iJGVD) 3.5KJPN; https://jmorp.megabank.tohoku.ac.jp) information. Finally, a novel heterozygous missense variant [NM_001844.5: c.2987G>A, (p. Gly996Asp)] of COL2A1 was found to be consistent with the diseased phenotype. None of the remaining variants were associated with skeletal disorders. This variant was not reported in any control genome database, such as the International Genome Sample Resource (IGSR; https://www.internationalgenome.org), the Human Genetic Variation Database (https://www.hgvd.genome.med.kyoto-u.ac.jp) or ToMMo (iJGVD). This variant was not reported in the Human Gene Mutation Database (https://www.hgmd.cf.ac.uk/ac/index.php) or LOVD-COL2A1 (https://databases.lovd.nl/shared/genes/COL2A1). This change results in a predicted substitution of glycine 996 with asparagine acid (p.Gly996Asp). The SIFT and PolyPhen2 scores were 0 and 0.999, respectively, confirming the deleterious effect of the missense variant on protein function.
To confirm the variant found in WES and whether the couple carried the same variant, we performed Sanger sequencing of the DNA from III-1, II-3, and II-6 using a previously reported method4. The missense variant was observed only in the affected fetus (III-1) and not in the couple (II-3 and II-6) (Fig. 2e). According to the American College of Medical Genetics and Genomics guidelines, the variant was considered “likely pathogenic (PS2+PM2+PP3+PP4)” 5. The fetus was diagnosed with ACG2 resulting from a novel missense variant of COL2A1.
As mentioned above in the couple’s inspection, this was a possible de novo variant. After informed consent was obtained, the female had a second spontaneous pregnancy and gave birth to a healthy child (III-2) with no symptoms of skeletal disorders.
ACG2 is an autosomal dominant fatal congenital skeletal disorder caused by defects in the COL2A1 gene1. COL2A1 is located on chromosome 12 and encodes a polypeptide chain of type 2 collagen. The triple-helical domain, which is the backbone of type 2 collagen, consists of a glycine-X-Y repeating motif. Glycine substitution in this domain causes ACG2, hypochondrogenesis, platyspondylic dysplasia (the Torrance type), SEDC, and SEMD (the Strudwick type)1,6. In this case, a substitution of glycine for asparagine acid was observed (p.Gly996Asp).
Prenatal diagnosis of skeletal disorders can be performed by fetal ultrasonography or computed tomography (CT)3. For postnatal diagnosis, radiographic examination and genetic analysis can be considered. In this case, fetal CT was not performed because of the early gestational period. The specific symptoms of ACG2 include fetal edema, marked limb shortening, a bell-shaped thorax, nonossification of the vertebral and pelvic bones, normal skull ossification, and internal rotation of the toes1,3. In our study, we observed a cystic hygroma and marked limb shortening in the fetus. There were some reports of cystic hygroma in ACG2, and the findings, in this case, were considered consistent with ACG2 7,8. Since the fetus was immature, ossification could not be evaluated3,9. Therefore, we performed WES to differentiate ACG2 from other skeletal disorders and found a novel missense variant of COL2A1. Based on the clinical findings and genetic analysis, we diagnosed the fetus with ACG2.
Somatic and germline mosaicism is also found in some parents of ACG2 patients7,10,11, which when present at low levels, are difficult to detect by Sanger sequencing using peripheral blood DNA samples. Therefore, if parents are suspected of having somatic or germline mosaicism, based on the family history, a close examination should be considered7,11.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3246.
References
Kannu, P., Bateman, J. & Savarirayan, R. Clinical phenotypes associated with type II collagen mutations. J. Paediatr. Child Health 48, E38–E43 (2012).
Mortier, G. R. et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. A 179, 2393–2419 (2019).
Sasahara, J. et al. Prenatal diagnosis of a rare skeletal dysplasia by three-dimensional computed tomographic imaging: Achondrogenesis type I. J. Jpn. Soc. Perinat. Neonatal Med. 50, 1336–1340 (2015).
Sato, T. et al. Novel TFAP2A mutation in a Japanese family with branchio-oculo-facial syndrome. Hum. Genome Var. 5, 5 (2018).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Terhal, P. A. et al. Mutation-based growth charts for SEDC and other COL2A1 related dysplasias. Am. J. Med. Genet. C Semin. Med. Genet. 160C, 205–216 (2012).
Forzano, F. et al. A familial case of achondrogenesis type II caused by a dominant COL2A1 mutation and “patchy” expression in the mosaic father. Am. J. Med. Genet. A 143A, 2815–2820 (2007).
Won, H. S. et al. A case of achondrogenesis type II associated with huge cystic hygroma: prenatal diagnosis by ultrasonography. Ultrasound Obstet. Gynecol. 14, 288–290 (1999).
Ulla, M. et al. Prenatal diagnosis of skeletal dysplasias: Contribution of three-dimensional computed tomography. Fetal Diagn. Ther. 29, 238–247 (2011).
Comstock, J. M. et al. Recurrence of achondrogenesis type 2 in sibs: Additional evidence for germline mosaicism. Am. J. Med. Genet. A 152A, 1822–1824 (2010).
Yamamoto, K. et al. Parental somatogonadal COL2A1 mosaicism contributes to intrafamilial recurrence in a family with type 2 collagenopathy. Am. J. Med. Genet. A 182, 454–460 (2020).
Acknowledgements
We are deeply grateful to the patient and their family members for participating in this study. This study was supported in part by a research grant from The General Insurance Association of Japan, Japan, The Jikei University Research Grant for the Career Development of Female Researchers with Children and the Jikei University Research Fund [grant number: 2021-006SR]. We would like to thank Editage (www.editage.com) for English language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kobayashi, Y., Ito, Y., Taniguchi, K. et al. Novel missense COL2A1 variant in a fetus with achondrogenesis type II. Hum Genome Var 9, 40 (2022). https://doi.org/10.1038/s41439-022-00218-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-022-00218-5
