
Abstract
We identified a three-generation Russian family with Lynch syndrome with a novel germline variant of the MSH6 gene. An 84-year-old female was diagnosed with endometrial adenocarcinoma at the age of 49 years. Her son was diagnosed with colorectal tubular adenoma at the age of 32 years. A germline nonsense variant (c.484 G > T:p.Gly162Ter) in exon 3 of the MSH6 gene was revealed by whole-exome sequencing. Sanger sequencing confirmed the cosegregation of the MSH6 nonsense variant in family members.
Similar content being viewed by others


Lynch syndrome (LS; OMIM#120435) is one of the manifestations of hereditary nonpolyposis colorectal cancer syndrome (HNPCC) and is associated with a genetic predisposition to different cancer types, including colon cancer, endometrial cancer, ovarian cancer, stomach cancer, and cancer of the small bowel, pancreas, and hepatobiliary tract1. In addition, data indicate a mildly increased risk of breast cancer2. HNPCC disorders show a proclivity to early onset and an excess of multiple primary tumors.
Dysfunction of DNA mismatch repair (MMR) leads to an increased frequency of biosynthetic errors generated during DNA replication and plays a critical role in the accumulation of mutations in cancer-related genes. LS results from a loss-of-function germline mutation in one of four different genes (MLH1, MSH2, MSH6, and PMS2) encoding mismatch repair proteins3. Mutations in the MMR genes MLH1, MSH2, MSH6, and PMS2 account for 40, 34, 18 and 8% of LS patients, respectively4. EPCAM deletion-associated hypermethylation of the MSH2 promoter varies in frequency between populations and may account for 10–40% of families with absent MSH2 protein in tumors5. A very small number of variants are in the more recently identified MSH36. The mismatch repair deficiency was shown to be frequently accompanied by microsatellite instability in tumors7. Two heterodimeric complexes, MSH2/MSH6 and MSH2/MSH3 (MutSα and MutSβ, respectively)8,9, are involved in mismatch recognition and initiation of repair10. MLH1 forms a complex with PMS2 and functions as an endonuclease.
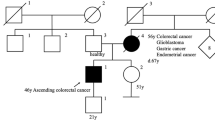
In this study, we describe a new pathogenic germline mutation in the MSH6 gene revealed by whole-exome sequencing (WES) in the genome of a proband of a three-generation family from northwestern Russia diagnosed with LS. An 84-year-old female who was diagnosed with endometrial adenocarcinoma at the age of 49 years was the proband (I) (Fig. 1). Between the ages of 49 and 79 years, the female patient developed an additional six primary malignant lesions: four were located in the intestine, and two were located in the breasts. She was successfully treated with surgery. Immunohistochemistry and mutational analysis showed that the lesions had different microsatellite stability, and the APC, KRAS, TGFBR2, TP53, PIC3CA, ARID1A, and e-cadherin status was not uniform among the lesions. Now, the affected family members include 2 individuals. Three members of the family (I, II, and III-1) provided peripheral blood samples and clinical information. The medical history was further investigated for disease occurrence. II is a 48-year-old man diagnosed with colorectal tubular adenoma of the sigmoid colon who underwent surgery at the age of 32 years. III-1 is a 21-year-old healthy female. The father of the proband died from colon cancer at the age of 52 years, and her mother died from breast cancer at the same age.

Asterisks depict the family members who provided blood samples. CRC colon cancer, BC breast cancer, EC endometrium adenocarcinoma, CoAd colorectal tubular adenoma of the sigmoid colon.
As we can see, the family history fulfilled the Amsterdam II criteria for LS: three family members were affected with LS-associated cancers, all of them were first-degree relatives, two generations were affected, and the proband was diagnosed before age 5011.
WES has enabled the identification of the gene responsible for LS in this family. WES of the blood sample from affected family member I was used by Genetico (Moscow, Russia). The Illumina NovaSeq 6000 platform (Illumina Inc., San Diego, CA, USA) was utilized for WES with a 100-bp paired-end run protocol. The WES results revealed several thousand synonymous and missense SNVs, deletions, insertions, duplications, and other variants in the proband’s genome. The variants of genes associated with the respective cancer predisposition syndrome, including Lynch syndrome, adenomatous polyposis and hamartomatous polyposis syndromes, hereditary breast-ovarian cancer, hereditary diffuse gastric cancer, Li-Fraumeni syndrome and other cancer-associated syndromes2,12,13,14, were searched for pathogenic mutations. Only variants with a minor allele frequency (MAF) < 0.01 in the Ensembl Project, ExAC browser, and gnomAD databases were included.
As a result, a stop-gain variant at chr2:28023059 G > T (GRCH37/hg19) in the mismatch repair gene MSH6 (NM_000179.3:c.484 G > T) was identified. This mutation is expected to result in termination of the amino acid chain after lysine-161 (p.Gly162Ter) in the N-terminal domain of the MSH6 protein. To our knowledge, the nonsense variant c.484 G > T in exon 3 of the MSH6 gene is absent from all population databases. It was not found in the Ensembl Project, ClinVar, or gnomAD, and it was not reported for LS previously. Only missense variant c.484 G > A:p.Gly162Arg with uncertain significance is present in ClinVar and gnomAD. As mismatch repair genes, including MSH6, are considered to be involved in tumorigenesis, the nonsense variant of MSH6, c.484 G > T, was subjected to further investigation.
This mutation was verified by Sanger sequencing (Fig. 2A). The segregation analysis of the mutation in the MSH6 gene among other family members was performed by PCR amplification and Sanger sequencing of targeted DNA fragments from two individuals, including one patient (II) and one unaffected family member (III-1). PCR products were sequenced by Genetico (Moscow, Russia). In addition to the proband, both family members analyzed (II and III-1) carried the nonsense variant MSH6: c.484 G > T (Fig. 2B and Fig. 2C, respectively).

A portion of the sequence chromatogram from MSH6 exon 3 demonstrates a heterozygous variant, c.484 G > T, in proband I (A), affected family member II (B) and unaffected family member III-1 (C). The reading in both directions is presented. The position of the nonsense mutation c.484 G > T [p.Gly162Ter] in the frame of exon 3 (D) and a schematic representation of the MSH6 protein (E) are shown.
The stop codon (Fig. 2D) eliminates almost all functional motifs: MSH2 interacting domain, MutS connector, core, clamp, and C-terminal domains (Fig. 2E). To date, it is known that c.467 C > G:p.S156Ter stop-gain mutation is associated with colon tumors, bladder tumors, and adenomatous polyps15. The S144I substitution causes inherited somatic deficiency in MMR, resulting in increased development of HNPCC16.
The ACMG/AMP classification17,18 for assessing the pathogenicity of different variants allowed us to estimate the nonsense variant (c.484 G > T) in exon 3 of MSH6. The null variant occurs in a gene where loss of function is a known mechanism of disease (PVS1)15; is absent in population databases (PM2) (allele frequency = 0 in Ensembl Project, ClinVar, and gnomAD); and cosegregates in affected members of the family (PP1) (this work). Moreover, Cancer Genome Interpreter (www.cancergenomeinterpreter.org) identified the MSH6 c.484 G > T variant as an oncogenic mutation. In summary, the nonsense variant (c.484 G > T) of MSH6 fulfilled the criteria of ACMG for a “pathogenic variant”, as one very strong (PVS1), one moderate (PM2), and one supportive (PP1) criteria of pathogenicity have been revealed for this germline variant.
Previous studies also reported nonsense variants of the MSH6 gene in Lynch syndrome families that were classified as deleterious: MSH6 c.1030 C > T:p.Gln344Ter encodes a protein with loss of 1017 amino acid residues19, pSer156Ter is associated with endometrial cancer; Gln177Ter is associated with LS, Ser200Ter is reported in LS or CRC, and Glu207Ter is associated with NPCRC or LSL in the HGMD; and Ser200Ter is reported in LS or CRC in gnomAD.
In conclusion, we reported a novel nonsense variant (c.484 G > T:p.Gly162Ter) in exon 3 of MSH6 (NM_000179.3). Clinical data, cosegregation analysis, and in silico prediction convincingly classified the MSH6 variant (c.484 G > T) as pathogenic and the loss-of-function mutation for LS in the family. Therefore, our results broaden the genotypic range of MSH6 mutations that cause LS. Visual invasive examinations, such as CT colonography, flexible sigmoidoscopy or colonoscopy every 1–2 years, were recommended to one unaffected family member (III-1) who also carried the MSH6 variant c.484 G > T.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3243.
References
Lynch, H. T., Lynch, J. F., Shaw, T. G. & Lubiński, J. HNPCC (Lynch Syndrome): differential diagnosis, molecular genetics and management - a review. Hered. Cancer Clin. Pr. 1, 7–18 (2003).
Porkka, N. K. et al. Does breast carcinoma belong to the Lynch syndrome tumor spectrum? − Somatic mutational profiles vs. ovarian and colorectal carcinomas. Oncotarget 11, 1244–1256 (2020).
Lynch, H. T., Snyder, C. L., Shaw, T. G., Heinen, C. D. & Hitchins, M. P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 15, 181–194 (2015).
Peltomäki, P. Update on Lynch syndrome genomics. Fam. Cancer 15, 385–393 (2016).
Ligtenberg, M. et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat. Genet 41, 112–117 (2009).
Duraturo, F. et al. Association of low-risk MSH3 and MSH2 variant alleles with Lynch syndrome: probability of synergistic effects. Int J. Cancer 129, 1643–1650 (2011).
Siegel, R., Naishadham, D. & Jemal, A. Cancer statistics 2013. CA Cancer J. Clin. 63, 11–30 (2013).
Arlow, T. et al. MutSα mismatch repair protein stability is governed by subunit interaction, acetylation, and ubiquitination. G3 11, jkaa065 (2021).
Warren, J. J. et al. Structure of the human MutSa DNA lesion recognition complex. Mol. Cell 26, 579–592 (2007).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Vasen, H. F., Watson, P., Mecklin, J. P. & Lynch, H. T. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative Group on HNPCC. Gastroenterology 116, 1453–1456 (1999).
Peltomӓki, P. & Vasen, H. Mutations associated with HNPCC predisposition – update of ICG-HNPCC/INSiGHT mutation database. Dis. Markers 20, 269–276 (2004).
Yurgelun, M. B. et al. Identification of a variety of mutations in cancer predisposition genes in patients with suspected Lynch syndrome. Gastroenterology 149, 604–613.e20 (2015).
Lagerstedt-Robinson, K. et al. Mismatch repair gene mutation spectrum in the Swedish Lynch syndrome population. Oncol. Rep. 36, 2823–2835 (2016).
Wijnen, J. et al. Familial endometrial cancer in female carriers of MSH6 germline mutations. Nat. Genet 23, 142–144 (1999).
Kariola, R. et al. MSH6 missense mutations are often associated with no or low cancer suceptibility. Br. J. Cancer 91, 1287–1292 (2004).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Jarvik, G. P. & Browning, B. L. Consideration of cosegregation in the pathogenicity classification of genomic variants. Am. J. Hum. Genet. 98, 1077–1081 (2016).
Pinto, C. et al. Co-occurrence of nonsense mutations in MSH6 and MSH2 in Lynch syndrome families evidencing that not all truncating mutations are equal. J. Hum. Genet. 61, 151–156 (2016).
Acknowledgements
We would like to thank all family members for their participation and cooperation.
Author information
Authors and Affiliations
Contributions
O.V. and V.V. conceived the study; E.M., G.B., A.S., and D.K. performed the experiments; V.V., Y.K., and D.K. performed the analysis. A.G., A.A., and Y.P. provided patient samples, V.V. and Y.K. drafted the manuscript. All authors have read and agreed to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Informed consent was obtained from all participating individuals, and the study protocol was approved by the Ethics Committee of Best Clinical Practice Company Ltd, St. Petersburg.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vostrukhina, O.A., Mirlina, E.D., Khmelkova, D.N. et al. An MSH6 germline pathogenic variant p.Gly162Ter associated with Lynch syndrome. Hum Genome Var 9, 37 (2022). https://doi.org/10.1038/s41439-022-00216-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-022-00216-7
