
Abstract
Here we report a consanguineous Pakistani family with multiple affected individuals with autosomal recessive congenital cataract (arCC). Exclusion analysis established linkage to chromosome 22q, and Sanger sequencing coupled with PCR-based chromosome walking identified a large homozygous genomic deletion. Our data suggest that this deletion leads to CRYBB2-CRYBB2P1 fusion, consisting of exons 1–5 of CRYBB2 and exon 6 of CRYBB2P1, the latter of which harbors the c.463 C > T (p.Gln155*) mutation, and is responsible for arCC.
Similar content being viewed by others

According to the Cat-Map database (https://cat-map.wustl.edu), 38 mutations identified in CRYBB2 are associated with autosomal dominant cataract (adC). CRYBB2 and its pseudogene CRYBB2P1 reside on chromosome 22, nearly 250 kb apart1. The gene conversion that shifts the NM_000496.3:c.463 C > T [p.(Gln155*)] variation from the pseudogene to CRYBB2 has been reported in different ethnic backgrounds segregating as an autosomal dominant trait (https://cat-map.wustl.edu).
A family (PKCC212) was included for a collaborative study to investigate the genetic basis of arCC. Institutional Review Board (IRB) approvals were obtained from Johns Hopkins University School of Medicine (Baltimore, MD), the National Institutes of Health (Bethesda, MD), and the National Centre of Excellence in Molecular Biology (Lahore, Pakistan). The study was completed in accordance with the Declaration of Helsinki, and all participants signed informed consent before enrollment.
Ophthalmic examinations, including slit-lamp microscopy, were performed at the Layton Rahmatulla Benevolent Trust (LRBT) Hospital (Lahore, Pakistan). Approximately 10 ml of blood was drawn from all subjects and stored in 50-ml Sterilin Falcon tubes with 20 mM EDTA. Genomic DNA was extracted, and exclusion analysis for reported arCC genes/loci using polymorphic short tandem repeat (STR) markers and Sanger sequencing were performed, as described2,3.
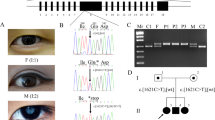
A total of eight individuals were enrolled in the study (Fig. 1a). The medical records of ophthalmic examinations confirmed that all three affected individuals had bilateral nuclear cataracts; surgery was performed on all the affected individuals except for individual V:3, before enrollment in the study (Fig. 1b). Importantly, unaffected family members exhibited no lens opacity, bilaterally. As shown in Supplementary Figs. 1 and 2, unaffected individuals IV:3 (age, 41 years) and V:1 (age, 16 years) had no signs of cataracts.

a Pedigree drawing of PKCC212 with the haplotypes of alleles for chromosome 22q microsatellite markers. Alleles forming the risk haplotype are shaded black, and alleles not cosegregating with arCC are shown in white. Square: male; circle: female; filled symbol: affected individual; the double line between individuals: consanguineous marriage; diagonal line through a symbol: deceased. b Slit-lamp photograph of affected individual V:3 in the PKCC212 showing nuclear cataracts.
Exclusion analysis confirmed linkage to chromosome 22q11.23, with a maximum two-point LOD score of 2.51 for marker D22S315 (Supplementary Table 1). Sanger sequencing excluded the possibility of pathogenic variants in CRYBA4, CRYBB1, CRYBB3, and in the first four coding exons (2–5) of CRYBB2. However, a primer pair specific for exon 6 of CRYBB2 failed to amplify, indicative of a genomic deletion and/or a rearrangement. We designed primer pairs to amplify the regions between CRYBB2 and LRP5L (BB2_LRP), exon 2 of LRP5L (LRP5L_Ex2), intron 3 of LRP5L (LRP5L_Int3), and a region of CRYBB2P1 (BB2P1_Int) (Supplementary Table 2). PCR failed to amplify all the abovementioned regions in affected individuals, but PCR products were obtained with genomic DNA from unaffected individuals i.e., III:3, IV:2, IV:3, V:1, and V:5 (Supplementary Fig. 3), suggesting a large genomic deletion.
In our effort to determine the extent of the deletion, we examined the PCR product of a forward primer annealing to CRYBB2 and a reverse primer annealing to CRYBB2P1 (Supplementary Table 2; BB2Int5_BB2P1Ex6), which theoretically would amplify an ~230-kb region, chr22:25230131-25459788 (GRCh38/Hg38). However, we obtained a PCR product of ~1800 bp (later determined to be 1832 bp by DNA sequencing) in all three affected individuals (Supplementary Fig. 4), suggesting that the genomic deletion eliminates nearly 228 kb DNA in members of PKCC212 harboring this deletion.
Sanger sequencing of the 1832 bp amplified PCR fragment identified 353 bp of CRYBB2 and 1145 of CRYBB2P1, whereas the 334-bp sequence was indistinguishable due to the high sequence similarity between CRYBB2 and CRYBB2P1 (Fig. 2 and Supplementary Data 1). Moreover, we identified the c.463 C > T variant in exon 6 of CRYBB2-CRYBB2P1, which leads to a premature stop codon at glutamine 155 (p.Gln155*) in the PKCC212 family (Supplementary Fig. 5). All three affected individuals were homozygous for the variant, whereas unaffected individuals were either heterozygous or homozygous for the wild-type allele (Fig. 1a). This deletion was not present in 96 unaffected individuals of Pakistani descent who were examined to rule out the presence of cataracts and 24 unaffected individuals of Saudi descent.
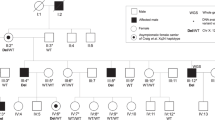
Taken together, the abovementioned data suggest that intron 5 of CRYBB2 became fused with intron 5 of CRYBB2P1, resulting in a hybrid CRYBB2-CRYBB2P1 gene consisting of the first five exons and a part of intron 5 of CRYBB2 followed by the remainder of intron 5, exon 6, and the 3’ UTR of CRYBB2P1 (Fig. 2).

a A 228-kb genomic deletion of chromosome 22q was identified in PKCC212. b The deletion in affected individuals of PKCC212 removes exon 6 of CRYBB2 (ENST00000651629.1), LRP5 L (ENST00000402859.6), and the first five exons of the CRYBB2 pseudogene (CRYBB2P1). Amplification results using the primer pair BB2Int5_BB2P1Ex6 (Supplementary Table 2) identified a 1832-bp fragment of chromosome 22q. Of this 1832-bp region, 353 and 1145 bp aligned with CRYBB2 and CRYBB2P1, respectively; 334 bp remained indistinguishable due to overwhelming similarity between CRYBB2 and CRYBB2P1. Note: The CRYBB2P1 transcripts, i.e., NR_ 033733.1 and NR_ 033734.1, consist of 5 and 6 exons, respectively. Exons 4 and 5 in the CRYBB2P1 transcripts NR_ 033733.1 and NR_ 033734.1 are homologous to CRYBB2 exon 6 harboring the p.Gln155* mutation. Note: Transcripts are as per Genome Reference Consortium Human Build 38 patch release 12 (GRCh38.p12) assembly.
We next investigated the whole exomes of the three affected individuals, i.e., individuals V:2, V:3, and V:4 of PKCC212, through next-generation sequencing. Whole-exome library preparation and next-generation sequencing of the affected individuals were performed commercially by Novogene Corporation Inc. (Davis, USA). The quality control analysis of exome data revealed >99% of the reads to be 150 base pairs, with 95% of the sequencing data yielding a PHRED score of 30 or above. High-throughput sequencing resulted in 48–66 million paired-end reads for each sample, and ~48–66 million reads (>99.8% of total reads) were uniquely mapped to the human genome (GRCh38.p13), representing an average 112× to 152× coverage for all three exomes (Supplementary Table 3). Lasergene Genomics Suite (DNASTAR, Madison, WI, USA) was used for reference-guided genome alignment and variant calling/annotation of the exome data. Paired-end raw reads were aligned to the human genome (GRCh38.p13), and the mapped reads were further processed for variant calling and annotation using DNASTAR (Madison, WI, USA) proprietary software (SeqMan NGen & ArrayStar Ver. 12) with default parameters. The stringent criterion was used to filter false-positive results from the potentially causal variants, as described3.
We first examined the linkage interval on chromosome 22q in the exome dataset, in particular, the 228-kb deletion. All variants (sequencing depth ≥5 reads) in the linkage interval were examined; however, we did not identify any variant originating from exon 6 of CRYBB2 or LRP5L in the affected individuals (Supplementary Data 2, 3, and 4). To rule out the possibility of a causative variant other than chromosome 22q deletion either within the linkage interval or elsewhere in the exome being responsible for the cataractous phenotype in PKCC212, all variants present in the three exomes were systematically filtered through an approach previously adopted to identify mutation in PEX53. Nevertheless, no variants in the exomes of individuals V:2, V:3, and V:4 justified the criterion for cataractogenesis (Supplementary Fig. 6).
Gene conversion resulting in the transfer of the c.463 C > T (p.Gln155*) variant from the pseudogene CRYBB2P1 to the sixth exon of CRYBB2 has been reported in multiple families with cataracts and manifests as an autosomal dominant trait4,5,6,7,8. In contrast, our data implicate a large genomic deletion encompassing exon 6 of CRYBB2, LRP5L, and most CRYBB2P1 responsible for arCC in PKCC212. The precise pathogenic mechanism of cataractogenesis due to the fusion of CRYBB2 and CRYBB2P1 remains unclear. It is noteworthy that the 3’UTR of hybrid CRYBB2 originates from the pseudogene that is not transcribed and/or translated and therefore most likely lacks the ability to protect nascent mRNA against degradation9. Given the importance of the 3’UTR in protecting mRNAs from degradation, it is tempting to speculate that the mRNA of the hybrid CRYBB2 (which includes the c.463 C > T (p.Gln155*) mutation) is not stable and is degraded. This would result in only wild-type mRNA being present in heterozygous carriers. We further speculate that a single functional CRYBB2 allele in heterozygous carriers is adequate for lens transparency. Conversely, individuals homozygous for the mutant allele completely lack functional CRYBB2, leading to a null phenotype and congenital cataracts.
Recently, Sun et al. reported a novel pathogenic mutation in LRP5L (c.107 C > G, p.P36R) in a congenital membranous cataract family10. Despite the possibility that concurrent deletion of LRP5L contributed to cataractogenesis in PKCC212, it is unlikely given the presence of null alleles in gnomAD database (https://gnomad.broadinstitute.org).
In conclusion, we identified a homozygous genomic deletion resulting in a hybrid CRYBB2 responsible for arCC. To the best of our knowledge, this is the first study reporting a large genomic deletion and subsequent fusion of CRYBB2-CRYBB2P1 being responsible for arCC. Mutations in all three β-crystallin genes, CRYBB1, CRYBB2, and CRYBB3, have been associated with arCC11,12, suggesting that haploinsufficiency of β-crystallins is tolerated by the ocular lens without affecting their respective physiological function in the lens.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3216.
References
Wistow, G. The human crystallin gene families. Hum. Genomics 6, 26 (2012).
Khan, S. Y. et al. Splice-site mutations identified in PDE6A responsible for retinitis pigmentosa in consanguineous Pakistani families. Mol. Vis. 21, 871 (2015).
Ali, M. et al. A missense allele of PEX5 is responsible for the defective import of PTS2 cargo proteins into peroxisomes. Hum. Genet. 140, 649–666 (2021).
Litt, M. et al. Autosomal dominant cerulean cataract is associated with a chain termination mutation in the human beta-crystallin gene CRYBB2. Hum. Mol. Genet. 6, 665–668 (1997).
Gill, D. et al. Genetic heterogeneity of the Coppock-like cataract: a mutation in CRYBB2 on chromosome 22q11.2. Invest. Ophthalmol. Vis. Sci. 41, 159–165 (2000).
Vanita et al. A unique form of autosomal dominant cataract explained by gene conversion between beta-crystallin B2 and its pseudogene. J. Med. Genet. 38, 392–396 (2001).
Yao, K. et al. Progressive polymorphic congenital cataract caused by a CRYBB2 mutation in a Chinese family. Mol. Vis. 11, 758–763 (2005).
Bateman, J. B. et al. Gene conversion mutation in crystallin, beta-B2 (CRYBB2) in a Chilean family with autosomal dominant cataract. Ophthalmology 114, 425–432 (2007).
Grzybowska, E., Wilczyńska, A. & Siedlecki, J. Regulatory functions of 3’UTRs. Biochem. Biophys. Res. Commun. 288, 291–295 (2001).
Sun, L. et al. The novel mutation P36R in LRP5L contributes to congenital membranous cataract via inhibition of laminin gamma1 and c-MAF. Graefes Arch. Clin. Exp. Ophthalmol. 258, 2737–2751 (2020).
Cohen, D. et al. Homozygous CRYBB1 deletion mutation underlies autosomal recessive congenital cataract. Invest. Ophthalmol. Vis. Sci. 48, 2208–2213 (2007).
Riazuddin, S. A. et al. Mutations in betaB3-crystallin associated with autosomal recessive cataract in two Pakistani families. Invest Ophthalmol. Vis. Sci. 46, 2100–2106 (2005).
Acknowledgements
The authors are grateful to all family members for their participation in this study. This study was supported in part by the King Khaled Eye Specialist Hospital-Johns Hopkins University collaboration grant (S.A.R.) and the National Eye Institute Grant 1R01EY022714 (S.A.R.).
Author information
Authors and Affiliations
Contributions
B.I., F.K., N.S., S.Y.K., B.R., S.R., J.F.H., and S.A.R. conceived of and designed the experiments. S.R., J.F.H., and S.A.R. contributed the reagents, materials, and analytical tools. B.I., F.K., N.S., S.Y.K., B.R., and M.A.N. performed the experiments. B.I., F.K., B.R., M.A.N., T.A.Q., S.R., J.F.H., and S.A.R. recruited the subjects. B.I., F.K., S.Y.K., B.R., S.R., J.F.R., and S.A.R. analyzed the data. B.I., F.K., N.S., B.R., S.Y.K., B.F., M.A.N., T.A.Q., S.R., J.F.H., and S.A.R. contributed to the writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Irum, B., Kabir, F., Shoshany, N. et al. A genomic deletion encompassing CRYBB2-CRYBB2P1 is responsible for autosomal recessive congenital cataracts. Hum Genome Var 9, 31 (2022). https://doi.org/10.1038/s41439-022-00208-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-022-00208-7
