
Abstract
In this study, we detected a novel pathogenic variant and a previously reported variant in RAB3GAP1 by whole-exome sequencing (NM_001172435.2: c.1552C>T, p.Gln518*; c.1471C>T, p.Arg491*). The first patient is a 3-year-old girl who presented with bilateral congenital cataracts, developmental delay, abnormal craniofacial features, drug-resistant constipation, and corpus callosum hypoplasia. The proband of the second family is a 13-year-old boy who suffers from developmental delay, quadriplegia, intellectual disability, abnormal craniofacial features, and corpus callosum hypoplasia.
Similar content being viewed by others

Warburg micro syndrome (WARBM) and Martsolf syndrome are rare genetic disorders caused by a deficiency in RAB18 protein (OMIM: #212720; #600118). Initially, they were regarded as separate entities; however, later, it was discovered that they are both caused by RAB18 deficiency1. Martsolf syndrome is the milder form of RAB18 deficiency, while Warburg micro syndrome is the more severe form.
Warburg micro syndrome is typically characterized by craniofacial, ophthalmic, genitourinary, skeletal anomalies, and neurologic disorders. Clinical findings in both syndromes are generally the same, with the former being milder than the latter1,2,3,4,5.
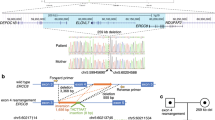
Here, we present two unrelated patients with WARBM1 caused by nonsense pathogenic variants (NM_001172435.2: c.1552C>, p.Gln518*; c.1471C>T, p.Arg491*) in RAB3GAP1, the first patients from Iran (Fig. 1A–D).

A The pedigree. B The electropherogram of the proband (IV-1), his father (III-5), and his mother (III-6). C The pedigree. D The electropherogram of the proband (IV-1), his father (III-5), and his mother (III-4).
We present the case of a 3-year-old Iranian girl, the fourth child of consanguineous parents. Their first child is a healthy 9-year-old boy. However, the second and third sons of the family did not survive infancy. The parents reported congenital cataracts and developmental delays in the deceased sons. They died at 5 and 8 months of age (however, it is not clear whether they died due to the same condition). The proband was born following a successful pregnancy at 37 weeks of gestation by caesarian section. At birth, her weight and head circumference were 2700 g and 32.5 cm, respectively. She had bilateral congenital cataracts, which were operated on at Day 35 after delivery. She had developmental delays in motor function, language and speech, and cognitive and social skills. The patient is not able to talk and also sit without support, but her hearing is intact. The patient has not gained weight appropriately. Currently, she weighs 8 kg. In physical examination, lower limb hyperreflexia was noted. She has not started speaking yet. In terms of abnormal craniofacial features, deep-set eyes, a prominent nasal root, a narrow mouth, and large anteverted ears can be recognized. The patient recently suffered from severe constipation and did not respond to medications (Fig. 2).

Deep-set eyes, a prominent nasal root, a narrow mouth, elbow contracture, and large anteverted ears are present.
Electroencephalography was not remarkable. Eye sonography revealed thick corneas, thin and cataractous lenses, mild irregularity of the inner surface of the posterior aspect of the globe wall, and mild elevation of the optic disc. Brain MRI shows abnormal signal alteration in the periventricular deep white matter with corpus callosum hypoplasia (Supplementary Fig. 1).
The results of routine biochemical tests, including liver, kidney, and thyroid function tests, were normal. TORCH screening test result at birth, complete blood count, and serum electrolytes were normal. The chromosomal analysis revealed a normal karyotype.
Whole-exome sequencing detected a novel nonsense pathogenic variant in RAB3GAP1 (NM_001172435.2: c.1552C>T, p.Gln518*), which was confirmed by Sanger sequencing.
The same variant was detected in her parents by Sanger sequencing.
The second proband is a 13-year-old Iranian boy, the only child of consanguineous parents (second cousins). He was born at term by normal vaginal delivery. Intrauterine growth restriction was recorded during gestation. At birth, he was 1900 g and 41 cm tall, and his head circumference was 31 cm. He was diagnosed with congenital cataracts. In infancy, he was hypotonic with poor head control. He currently has severe spastic quadriplegia. His language acquisition is minimal. He is in the first Tanner stage of pubertal development. He cannot communicate effectively with his parents and caregivers. Sonographic findings were similar to those of the proband of Family 1. Abnormal craniofacial features in the proband include deep-set eyes, a prominent nasal bridge and root, a maxillary protrusion, and large anteverted ears. Failure to thrive was present during his development course, which did not respond to interventions (Figs. 1C, 3).

Deep-set eyes, a prominent nasal root, a narrow mouth, a maxillary protrusion, elbow contracture, and large anteverted ears are present.
Congenital diaphragmatic hernia (CDH) was detected prenatally and confirmed after delivery. His CDH was repaired on the second day of life. He quickly recovered from the surgery. Lens replacement surgery was conducted when he was six months old. Posterior vitreous detachment was noticed following the surgery. Following the surgery, his sight deteriorated to no light perception.
He had a micropenis, and his testes were not palpable at birth. Increased dehydroepiandrosterone sulfate and decreased luteinizing hormone, follicle-stimulating hormone, and testosterone were recorded in the following months. Sonography was unsuccessful in localizing the undescended testes. Laparoscopic orchiopexy was carried out at 18 months of age; however, the operation was not successful because the testes were not present in the abdominal cavity. Due to abnormal kidney function, he received kidney transplantation at four years of age.
Brain magnetic resonance imaging showed CSF prominence suggesting cerebral atrophy and hypoplasia of the body and splenium of the corpus callosum. Electroencephalography following a few seizure-like episodes at 6 years of age (mild clonus at the right shoulder) failed to show any epileptic activity except in the low-amplitude background (Supplementary Figs. 1D, 2, 3).
TORCH infections were ruled out. Neonatal screening and testing for metabolic disorders were carried out during the first six months of life. No metabolic disorder was detected. He has normal thyroid function. Whole-exome sequencing was conducted for the proband. We found that the proband was homozygous for a variant in RAB3GAP1 (NM_001172435.2: c.1471C>T, p.Arg491*; rs752667359). The presence of the variant was confirmed by Sanger sequencing. Sanger sequencing detected the same variant in his parents.
None of the variants presented in this study are reported in the gnomAD, Iranome, GME, or TogoVar databases. While p.Arg491* was registered in the ClinVar database, p.Gln518* is novel. p.Gln518* is classified as pathogenic by BayesDel addAF, BayesDel noAF, DANN, EIGEN, EIGEN PC, FATHMM-MKL, FATHMM-XF, LRT, and MutationTaster. Using BayesDel addAF, BayesDel noAF, DANN, EIGEN, EIGEN PC, FATHMM-MKL, LRT and MutationTaster, Arg491* was categorized as pathogenic. Both variants are categorized as pathogenic based on the PVS1, PM2, PP3, and PP5 ACMG classification criteria.
Warburg micro syndrome is a rare autosomal-recessive disorder primarily affecting the visual, nervous, and endocrine systems. Warburg micro syndrome was first described in a Pakistani family presenting with microcornea, microcephaly, congenital cataracts, severe mental retardation, retinal dystrophy, optic nerve atrophy, hypothalamic hypogenitalism, and corpus callosum agenesis6. One of the variants detected in the current study was previously described by Morris-Rosendahl et al. in a Turkish patient7. Comparing the phenotype of proband 2 of the current study with the previously reported patient, the regressive nature of the development in the patient presented becomes more evident.
RAB18 deficiency was assumed to be the underlying mechanism of Martsolf and Warburg micro syndromes. RAB proteins are a large family of GTPase enzymes that facilitate cell tracking. RAB GTPases typically connect to membranes by binding two carboxy-terminal cysteine residues using their hydrophobic geranylgeranyl groups. Prenylation facilitates their role in synchronizing membrane trafficking8. RAB proteins have been shown to have a diverse set of roles in human disorders such as immunodeficiency, cancer, and neurodegenerative disorders8,9. Rab18 deficiency can arise from mutations in any of the following genes: RAB3GAP1, RAB3GAP2, RAB18, or TBC1D20. Seventy-five percent of RAB18 deficiency cases were attributed to mutations in RAB3GAP11.
In this paper, we present the first Iranian patients with pathogenic variants in RAB3GAP1 causing Warburg micro syndrome. In 2020, Hossein Hozhabri et al. reported two Iranian siblings with Martsolf syndrome caused by a pathogenic variant in TBC1D2010. The cases described in the study by Hossein Hozhabri et al. and those in the present study are the only RAB18 deficiency cases reported from Iran. These reports suggest that RAB18 deficiency may be underdiagnosed in Iran, requiring a more rigorous approach toward the molecular diagnosis of children with signs and symptoms typical of genetic diseases, including Martsolf and Warburg micro syndrome. Compared with previous reports, one of our patients suffers from severe drug-resistant constipation, which was not mentioned in the literature.
To date, among the 69 different mutations of various types identified in RAB3GAP1 in 65 families, no mutation hotspot has been recognized. This implies that all 24 exons in each patient have to be studied1,11,12. Molecular diagnosis would considerably benefit patients and caregivers. Molecular diagnosis would permit genetic counseling for future children of the family. Furthermore, the clinical course of the disease in the proband, despite the phenotypic variabilities, becomes predictable. Thus, clinical interventions for signs and symptoms such as cryptorchidism and developmental delay can be beneficial.
HGV Database
The relevant data from this DataReport are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3097, https://doi.org/10.6084/m9.figshare.hgv.310.
Availability of data and material
All data generated or analyzed during this study are included in the final published article.
References
Handley M., Sheridan E. RAB18 Deficiency. GeneReviews® (Seattle Univ Washington, Seattle; 1993-2021). 2020;1–23.
Gagnier, J. J. et al. The CARE guidelines: consensus-based clinical case reporting guideline development. Glob. Adv. Heal Med. 2, 38–43 (2013).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010). 2010/01/15.
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164–e164 (2010).
Rychlik, W. OLIGO 7 primer analysis software. Methods Mol. Biol. 402, 35–60 (2007).
Warburg, M., Sjö, O., Fledelius, H. C. & Pedersen, S. A. Autosomal recesssive microcephaly, microcornea, congenital cataract, mental retardation, optic atrophy, and hypogenitalism: micro syndrome. Am. J. Dis. Child. 147, 1309–1312 (1993).
Morris-Rosendahl, D. J. et al. New RAB3GAP1 mutations in patients with Warburg Micro Syndrome from different ethnic backgrounds and a possible founder effect in the Danish. Eur. J. Hum. Genet. 18, 1100–1106 (2010).
Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. cell Biol. 10, 513–525 (2009).
Zerial, M. & McBride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. cell Biol. 2, 107–117 (2001).
Hozhabri, H., Talebi, M., Mehrjardi, M. Y. V., De Luca, A. & Dehghani, M. Martsolf syndrome with novel mutation in the TBC1D20 gene in a family from Iran. Am. J. Med. Genet. A. 182, 957–961 (2020).
Handley, M. T. et al. Mutation spectrum in RAB3GAP1, RAB3GAP2, and RAB18 and genotype–phenotype correlations in Warburg micro syndrome and Martsolf syndrome. Hum. Mutat. 34, 686–696 (2013).
Aligianis, I. A. et al. Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat. Genet. 37, 221–224 (2005).
Acknowledgements
The authors would like to thank Comprehensive Medical Genetics Center, Shiraz, Fars province, Iran, for their invaluable contribution to this study.
Author information
Authors and Affiliations
Contributions
This study was designed by S.Z., O.A., and H.J.K. Sanger and whole-exome sequencing was carried out by S.M.P., H.J.K., S.A.D., and S.M.B.T. Physical examination was performed by O.A., M.D., and A.F. The final manuscript was written and edited by M.D., S.Z., and A.F. All contributing authors approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participants
Written informed consent was obtained from each patient or, in the case of minors, from their parents. This study was approved by the Ethics Committee of Shiraz University of Medical Sciences.
Consent for publication
Consent for publication was obtained from the patient.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alavi, O., Khamirani, H.J., Zoghi, S. et al. Two novel Warburg micro syndrome 1 cases caused by pathogenic variants in RAB3GAP1. Hum Genome Var 8, 39 (2021). https://doi.org/10.1038/s41439-021-00171-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-021-00171-9

{kind=link}
{kind=link}