
Abstract
Hyperprolinemia type I (HPI) is an autosomal recessive metabolic disorder caused by defects in proline oxidase. We herein describe a case of a patient with HPI and harboring the NM_016335.4 (PRODH_v001):c.1397 C > T (p.T466M) mutation and polymorphisms in the PRODH gene, as detected by plasma amino acid analysis and Sanger sequencing. The patient presented with short stature, carbohydrate-rich dietary preferences, and mild intellectual disability that was suggestive of a neurodevelopmental or learning disorder.
Similar content being viewed by others

Hyperprolinemia type I (HPI) is an autosomal recessive metabolic disorder caused by defects in proline oxidase (POX, EC: 1.5.99.8), also called proline dehydrogenase (PRODH). The PRODH gene is located on chromosome 22 (22q11.21) and encodes the POX enzyme, which converts proline to pyrroline-5-carboxylate (P5C) in mitochondria.
The clinical phenotype of HPI has not been clearly characterized. Patients with HPI may present with seizures, intellectual disability, language delay, autism spectrum disorder (ASD), schizophrenia, and/or bipolar disorder. Conversely, others are asymptomatic1,2,3. Very few case reports of patients with HPI have been reported worldwide, with only two described in Japan4,5.
An 8-year-old boy was referred to our institution for further investigation of the cause of his short stature and suspected hypoglycemia. He was the third child of healthy nonconsanguineous parents. He was born without complications at 40 weeks of gestation, with a length of 50 cm (+0.3 SD) and a weight of 2932 g (−0.9 SD). He was able to walk independently at the age of 1 year and 2 months; speech milestones were reached at the age of 2 years. He had certain food preferences; for example, although the amount of food he ingested was normal, he tended to prefer carbohydrate-rich foods such as boiled rice and disliked a protein-rich diet including meat and dairy. He had no significant past medical history; however, an incidental finding of short stature (89.7 cm [−2.0 SD]) was noted during a routine medical examination at 3 years and 6 months. The trend persisted, and at the age of 5 years, low fasting blood insulin-like growth factor-1 levels (52 ng/mL) were detected, suggesting growth hormone (GH) deficiency. He underwent further investigation to evaluate hypothalamic-pituitary function; clonidine stimulation, arginine infusion, insulin-thyrotropin-releasing hormone/luteinizing hormone-releasing hormone, L-dopa, GH-releasing peptide-2, and Fishberg concentration tests were all normal. At the age of 7 years, he developed frequent vomiting, and was a hypoglycemic attack was suspected. Nevertheless, he presented fasting normoglycemia (91 mg/dL) at admission, and his adrenal function test was normal. Acylcarnitine profile analysis revealed no abnormalities. However, plasma amino acid analysis detected high proline levels (530 μmol/L; reference: 78 − 273 μmol/L) (Table 1A). An intelligence assessment using the Wechsler Intelligence Scale for Children-Fourth Edition (WISC-IV) at the age of 6 years and 3 months revealed the following: a score of 83 in Full Scale IQ (FSIQ), 80 in Verbal Comprehension Index (VCI), 115 in Perceptual Reasoning Index (PRI), 68 in Working Memory Index (WMI), and 78 in Processing Speed Index (PSI). These characteristics suggest a tendency toward neurodevelopmental disorders, including autism, attention-deficit hyperactivity disorder, and learning disorders.
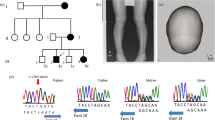
Upon referral to our team, we further investigated the patient’s developmental history. His mother reported that he could not keep up with regular class studies in primary school or perform detailed work. He had difficulty in reading and writing Kanji. WISC-IV intelligence assessment at 9 years and 9 months was 73, 86, 91, 63 and 67 for FSIQ, VCI, PRI, WMI, and PSI, respectively, indicating mild intellectual disability. Neurological examination revealed slightly poor coordination, but there was no history of impulsive behavior. Brain magnetic resonance imaging (MRI) (Fig. 1A) and magnetic resonance spectroscopy revealed nonspecific findings, and his electroencephalogram was unremarkable. However, Sanger sequencing detected the homozygous mutation c.1397 C > T (p.T466M) in the PRODH gene as well as some homozygous variants (Fig. 1B and Table 1B), confirming the diagnosis of HPI. No pathogenic variants were detected in ALDH4A1, which encodes P5C dehydrogenase (EC 1.2.1.88) (data not shown), defects of which cause hyperprolinemia type II. At the age of 10 years and 3 months, the patient was doing well in a supported education class and did not require medication. Written informed consent was obtained from the patient’s parents for this report. This study was approved by the Institutional Ethics Committee of the Faculty of Life Science, Kumamoto University.

A Brain MRI (T2-weighted image). B Sanger sequencing. The patient’s father and mother carried a heterozygous mutation of c.1397 C > T (p.T466M) in the PRODH gene. C The growth curve of the patient from birth. Filled circles, height measurements; open circles, weight measurements. These circles are superimposed onto the Cross-sectional Growth Chart for Boys (0–18 y) provided by the 2000 National Growth Survey on Preschool Children & School Health Statistics Research. The height of our patient was −2.0 SD of the mean height for his age among Japanese male children. The dotted lines at −2.5 SD and −3.0 SD of height indicate the criteria for starting growth hormone (GH) treatment for GH insufficiency and achondroplasia. SD: standard deviation.
In Table 1C, we summarize nine cases, including ours, of the POX p.T466M variant1,6,7. The PRODH gene, which is located in the 22q11 chromosomal region, is hemideleted in 22q11 deletion syndrome, also known as velo-cardio-facial syndrome (VCFS)8. VCFS shares some clinical features with HPI.
A combination of mutations and polymorphisms in the PRODH gene cause dysregulation of POX enzyme activity and may lead to a variety of phenotypes in HPI, including neuronal function disorders. The proline metabolic pathway links cellular proline levels with glutamate and glutamine levels in neurons, and POX has been proposed to play a regulatory role in glutamatergic neurotransmission by affecting the cellular concentration of glutamate9. Moreover, proline is thought to induce oxidative stress in the rat brain10,11. Hyperprolinemia induces significant oxidative damage to proteins, lipids, and DNA12, decreases the activities of antioxidant enzymes, and induces lipid peroxidation in the blood of rats13.
Mouse models of POX deficiency, which is also present in individuals with schizophrenia, exhibit increases in neurotransmitter release at glutamatergic synapses as well as deficits in associative learning and response to psychomimetic drugs14. Furthermore, hyperprolinemia has been suggested to be a risk factor for schizophrenia14,15, and polymorphisms in PRODH, such as rs372055, which this case harbored, are thought to correlate with schizophrenia16.
Some pharmacological, biochemical, and behavioral studies have suggested the involvement of the glutamatergic system in ASD pathology17, and ASD has been considered a common clinical manifestation of HPI1,2,6. Poor social, adaptive, and academic skills are often evident in patients with HPI3. Our patient displayed a complex learning disorder, including deficits in reading, writing, and math, and required specific educational training. Although this case and other reports suggest a correlation between intelligence and plasma proline levels6,8, further studies are required for confirmation.
Bender et al. proposed the following classification of PRODH mutations based on POX activity reduction: mild (<30%), moderate (30–70%), and severe (>70%)18. POX uses flavin adenine dinucleotide (FAD) as a cofactor. The homozygous c.1397 C > T (p.T466M) mutation results in reduced enzyme activity to less than 20% of the control18. T466 in POX interacts with the adenine moiety of FAD to stabilize noncovalent binding of the cofactor to the POX apoenzyme, and the T466M mutation alters the affinity of the POX apoenzyme for FAD.
Genotype-phenotype correlations in HPI have been suggested2,6. For example, Rosa et al.3 reported two patients with the same PRODH genotype and the same range of plasma proline levels (376 and 493 μmol/L); these patients presented with a similar neurobehavioral profile, including aggressiveness and sexual disinhibition. Our patient first presented with short stature and mild hyperprolinemia but without obvious intellectual disability, though developmental delay became more noticeable with age. Alexandra et al.1 described a patient with the p.T466M variant who presented with clinical manifestations and blood proline levels similar to those in our case. Although p.T466M is predicted to be damaging (0.943) in Polyphen 2 (http://genetics.bwh.harvard.edu/pph2/), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar) provides conflicting interpretations of its pathogenicity (Table 1B). Therefore, the clinical impact of p.T466M remains unclear. Although short stature has not been reported as a clinical manifestation of HPI, our case showed short stature persisting from infancy (Fig. 1C). Harries et al.19 reported persistent short stature until the age of 27 months in a patient with HPI receiving a low-proline diet. Moreover, van de Ven et al.20 described a patient with HPI presenting with a variable eating behavior pattern that is consistent with that of our patient. Indeed, our patient preferred a carbohydrate-rich diet and disliked protein-rich foods. The manifestations of short stature and food preference may be derived from HPI, but more case reports are needed to clarify this association.
In conclusion, we encountered a case of HPI that was first detected through plasma amino acid analysis performed during the detailed evaluation of short stature in a child. Mild intellectual disability, mild learning disorders, autism tendencies, and attention-deficit hyperactivity disorder (ADHD) tendencies are considered phenotypes related to HPI. The patient’s unique dietary habit is also thought to be one of the phenotypes of HPI, and blood proline levels vary depending on the dietary content. As very few cases of HPI have been reported to date, other patients may display as-yet-unidentified phenotypes. The accumulation of more cases is essential to further our understanding of the clinical characteristics of HPI.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at
https://doi.org/10.6084/m9.figshare.hgv.3045
https://doi.org/10.6084/m9.figshare.hgv.3048
https://doi.org/10.6084/m9.figshare.hgv.3051
https://doi.org/10.6084/m9.figshare.hgv.3054
https://doi.org/10.6084/m9.figshare.hgv.3057
https://doi.org/10.6084/m9.figshare.hgv.3060
https://doi.org/10.6084/m9.figshare.hgv.3063
References
Afenjar, A. et al. Early neurological phenotype in 4 children with biallelic PRODH mutations. Brain Dev. 29, 547–552 (2007).
Di Rosa, G. et al. Type I hyperprolinemia and proline dehydrogenase (PRODH) mutations in four Italian children with epilepsy and mental retardation. Psychiatr. Genet. 18, 40–42 (2008).
Di Rosa, G., Nicotera, A. G., Lenzo, P., Spanò, M. & Tortorella, G. Long-term neuropsychiatric follow-up in hyperprolinemia type I. Psychiatr. Genet. 24, 172–175 (2014).
Oyanagi, K. et al. Clinical, biochemical and enzymatic studies in type I hyperprolinemia associated with chromosomal abnormality. Tohoku J. Exp. Med. 151, 465–475 (1987).
Ishikawa, Y. et al. A case of type I hyperprolinemia associated with photogenic epilepsy (in Japanese). No Hattatsu (Tokyo) 23, 81–86 (1991).
Guilmatre, A. et al. Type I hyperprolinemia: genotype/phenotype correlations. Hum. Mutat. 31, 961–965 (2010).
Chérot, E. et al. Using medical exome sequencing to identify the causes of neurodevelopmental disorders: experiences of 2 clinical units and 216 patients. Clin. Genet. 93, 567–576 (2018).
Raux, G. et al. Involvement of hyperprolinemia in cognitive and psychiatric features of the 22q11 deletion syndrome. Hum. Mol. Genet. 16, 83–91 (2007).
Cappelletti, P. et al. Proline oxidase controls proline, glutamate, and glutamine cellular concentrations in a U87 glioblastoma cell line. PLoS ONE 13, e0196283 (2018).
Delwing, D. et al. Proline induces oxidative stress in cerebral cortex of rats. Int J. Dev. Neurosci. 21, 105–110 (2003).
Delwing, D. et al. In vivo and in vitro effects of proline on some parameters of oxidative stress in rat brain. Brain Res. 991, 180–186 (2003).
Ferreira, A. G. et al. Hyperprolinemia induces DNA, protein and lipid damage in blood of rats: antioxidant protection. Int J. Biochem. Cell Biol. 54, 20–25 (2014).
Roecker, R. et al. Proline alters antioxidant enzyme defenses and lipoperoxidation in the erythrocytes and plasma of rats: in vitro and in vivo studies. Biol. Trace Elem. Res. 147, 172–179 (2012).
Paterlini, M. et al. Transcriptional and behavioral interaction between 22q11.2 orthologs modulates schizophrenia-related phenotypes in mice. Nat. Neurosci. 8, 1586–1594 (2005).
Clelland, C. L. et al. Evidence for association of hyperprolinemia with schizophrenia and a measure of clinical outcome. Schizophr. Res. 131, 139–145 (2011).
Guo, X., Tang, P., Yang, C. & Li, R. Proline dehydrogenase gene (PRODH) polymorphisms and schizophrenia susceptibility: a meta-analysis. Metab. Brain Dis. 33, 89–97 (2018).
Choudhury, P. R., Lahiri, S. & Rajamma, U. Glutamate mediated signaling in the pathophysiology of autism spectrum disorders. Pharm. Biochem. Behav. 100, 841–849 (2012).
Bender, H. U. et al. Functional consequences of PRODH missense mutations. Am. J. Hum. Genet. 76, 409–420 (2005).
Harries, J. T., Piesowicz, A. T., Seakins, J. W., Francis, D. E. & Wolff, O. H. Low proline diet in type I hyperprolinaemia. Arch. Dis. Child 46, 72–81 (1971).
Van De Ven, S. et al. Long-term clinical outcome, therapy and mild mitochondrial dysfunction in hyperprolinemia. J. Inherit. Metab. Dis. 37, 383–390 (2014).
Acknowledgements
We thank the patient’s parents for their support and all the medical staff involved in treating the patient. This study was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (Japan Society for the Promotion of Science [JSPS] KAKENHI: grant number JP20K08207).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hama, R., Kido, J., Sugawara, K. et al. Hyperprolinemia type I caused by homozygous p.T466M mutation in PRODH. Hum Genome Var 8, 28 (2021). https://doi.org/10.1038/s41439-021-00159-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-021-00159-5
This article is cited by
-
Genetic components of microdeletion syndromes and their role in determining schizophrenia traits
Molecular Biology Reports (2024)
