
Abstract
A recurrent ZSWIM6 variant, NM_020928.2:c.2737C>T [p.Arg913*], was identified in a Japanese male patient with severe neurodevelopmental delay, epilepsy, distinctive facial features, microcephaly, growth deficiency, abnormal behavior, and frequent vomiting but without frontonasal or limb malformations. In this patient, distinctive facial features gradually became apparent with age, and severe vomiting caused by gastroesophageal reflux continued even after percutaneous endoscopic gastrostomy.
Similar content being viewed by others


In 2014, a de novo variant in the zinc finger SWIM-type containing 6 gene (ZSWIM6) was commonly identified in four patients with acromelic frontonasal dysostosis (MIM #603671)1, a rare disorder characterized by distinct craniofacial, brain, and limb malformations, including frontonasal dysplasia, interhemispheric lipoma, agenesis of the corpus callosum, tibial hemimelia, preaxial polydactyly of the feet, and intellectual disability2. In 2017, a different ZSWIM6 nonsense variant was identified in six patients with severe intellectual disability without frontonasal or limb malformations (MIM #617865)3. This evidence indicates that ZSWIM6 has a clear genotype–phenotype correlation. Recently, we identified the same nonsense variant in a Japanese patient with severe neurodevelopmental delay but without frontonasal or limb malformations.
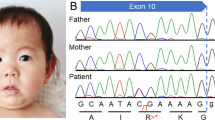
The male patient is 14 years old. He was born with a birth weight of 2720 g (mean), a length of 47.5 cm (10th–25th centile), and an occipitofrontal circumference (OFC) of 33.0 cm (mean) to healthy parents (a 33-year-old father and a 28-year-old mother). Soon after birth, a heart murmur was detected, and echocardiography showed a small atrial septal defect that spontaneously closed. At 5 months of age, psychomotor developmental delay was noted. At 2 years, he was examined for frequent vomiting, and gastroesophageal reflux (GER) was detected. At that time, he could still not control the movement of his head, and apparent muscular hypotonia was noted. Brain magnetic resonance imaging was performed, but no definite abnormality was identified other than the cavity of the septum pellucidum. At 6 years, he started to show autistic behaviors, including self-injury. He often presented with insomnia. At 12 years, he started to experience generalized seizures. Although there were no paroxysmal discharges on electroencephalography, his epileptic attacks were intractable, despite the combined use of antiepileptic drugs. Owing to frequent vomiting, he underwent percutaneous endoscopic gastrostomy at 12 years; however, he continuously experienced frequent vomiting. Over time, he began to show spasticity in his lower extremities. At present, his height is 130 cm (<3rd centile), his weight is 17.3 cm (<3rd centile), and his OFC is 47.5 cm (<3rd centile), indicating severe growth deficiency and microcephaly. He showed distinctive facial features with right internal strabismus, which were not noted during the infantile period (Fig. 1A). He also showed severe psychomotor developmental delay and had no head control, and he was incapable of meaningful speech. His maintenance of eye contact was poor. Although insomnia was controlled by the prescription of melatonin, his frequent vomiting remained intractable.

A No distinctive features are noted in infancy. B Distinctive features, including a prominent forehead, prominent supraorbital ridges, and prominent cheeks, are shown at 14 years. These photos were provided by his family with written informed consent. C Results of Sanger sequencing. The patient shows a de novo variant (C>T).
Prior conventional chromosomal G-banding showed a normal male karyotype of 46XY, and subsequently performed chromosomal microarray testing performed as described previously showed no abnormal findings4. Thus, this patient was enrolled in the research project “Initiative on Rare and Undiagnosed Diseases (IRUD)”5,6. This study was performed in accordance with the Declaration of Helsinki and was approved by the ethics committee of our institution. After obtaining written informed consent, we collected blood samples from the patient and both parents. Genomic DNA was extracted from the peripheral blood of the individuals using a standard protocol. Exome sequencing was performed using trio samples, including parental samples, as described previously7. Finally, we identified a heterozygous variant, NM_020928.2(ZSWIM6):c.2737C>T [p.Arg913*], the same variant reported by Palmer et al.3. Because neither parent showed this variant, it was considered de novo. Standard PCR-Sanger sequencing confirmed the de novo occurrence of this variant (Fig. 1B). No other variants that might be related to the clinical features of this patient could be identified. The clinical features of the present patient are summarized in Table 1, together with the clinical data provided by Palmer et al.3. As shown, the patient showed clinical features that were similar to those of the other patients. Therefore, we determined that the identified ZSWIM6 variant was responsible for his clinical features.
ZSWIM6 is located on chromosome 5q12.1 and consists of 14 exons. Using mice, it was revealed that Zswim6 is initially expressed widely during embryonic brain development, suggesting a critical role of this gene in neuronal development8. There are some reports of chromosomal microdeletions in the ZSWIM6 region9,10; however, patients with microdeletions involving ZSWIM6 show milder neurodevelopmental disorders than patients with the p.Arg913* ZSWIM6 variant. This indicates that the haploinsufficiency of ZSWIM6 is not a mechanism. The p.Arg913* ZSWIM6 variant lies within the penultimate exon 13, 48 bp upstream of the last exon/exon junction, suggesting that the ZSWIM6 mRNA encoding this pretermination codon may escape nonsense-mediated decay. Thus, it is considered that the p.Arg913* ZSWIM6 variant may result in a dominant-negative effect due to the production of a truncated ZSWIM6 protein that lacks the Sin-3-like domain in exon 14.
The present patient showed no distinctive facial features during infancy; however, his facial appearance changed with age, and the characteristics gradually appeared, as shown in Fig. 1A. This finding is also common with patients reported by Palmer et al.3. The most prominent symptom in the present patient was frequent vomiting. Palmer et al. reported that 4/7 patients (57%) showed GER, and two of them received gastrostomy3. Similarly, the present patient also underwent percutaneous endoscopic gastrostomy; however, severe vomiting persisted. Therefore, we are currently considering the placement of an elemental diet tube into the duodenum to avoid vomiting. It is unknown why patients with the p.Arg913* ZSWIM6 variant show such severe GER. The mechanism underlying severe GER and an appropriate treatment approach should be identified in the future.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.2999.
References
Smith, J. D. et al. Exome sequencing identifies a recurrent de novo ZSWIM6 mutation associated with acromelic frontonasal dysostosis. Am. J. Hum. Genet. 95, 235–240 (2014).
Twigg, S. R. et al. Acromelic frontonasal dysostosis and ZSWIM6 mutation: phenotypic spectrum and mosaicism. Clin. Genet. 90, 270–275 (2016).
Palmer, E. E. et al. A recurrent de novo nonsense variant in ZSWIM6 results in severe intellectual disability without frontonasal or limb malformations. Am. J. Hum. Genet. 101, 995–1005 (2017).
Yamamoto, T., Shimojima, K., Ondo, Y., Shimakawa, S. & Okamoto, N. MED13L haploinsufficiency syndrome: a de novo frameshift and recurrent intragenic deletions due to parental mosaicism. Am. J. Med. Genet. A 173, 1264–1269 (2017).
Adachi, T. et al. Japan’s initiative on rare and undiagnosed diseases (IRUD): towards an end to the diagnostic odyssey. Eur. J. Hum. Genet. 25, 1025–1028 (2017).
Adachi, T. et al. Survey on patients with undiagnosed diseases in Japan: potential patient numbers benefiting from Japan’s initiative on rare and undiagnosed diseases (IRUD). Orphanet J. Rare Dis. 13, 208 (2018).
Yamamoto-Shimojima, K. et al. Elucidation of the pathogenic mechanism and potential treatment strategy for a female patient with spastic paraplegia derived from a single-nucleotide deletion in PLP1. J. Hum. Genet. 64, 665–671 (2019).
Tischfield, D. J. et al. Loss of the neurodevelopmental gene Zswim6 alters striatal morphology and motor regulation. Neurobiol. Dis. 103, 174–183 (2017).
Jaillard, S. et al. 5q12.1 deletion: delineation of a phenotype including mental retardation and ocular defects. Am. J. Med. Genet. A 155a, 725–731 (2011).
Shimizu, T. et al. Fine-mapping of 5q12.1-13.3 unveils new genetic contributors to caries. Caries Res. 47, 273–283 (2013).
Acknowledgements
We appreciate the cooperation of the patient and his parents for this study. This work was supported by the Initiative on Rare and Undiagnosed Diseases (grant number 20ek0109301) from the Japan Agency for Medical Research and Development (AMED).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yanagishita, T., Eto, K., Yamamoto-Shimojima, K. et al. A recurrent de novo ZSWIM6 variant in a Japanese patient with severe neurodevelopmental delay and frequent vomiting. Hum Genome Var 8, 16 (2021). https://doi.org/10.1038/s41439-021-00148-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-021-00148-8
This article is cited by
-
Entry of ZSWIM4 to the nucleus is crucial for its inhibition of KIT and BMAL1 in gastrointestinal stromal tumors
Cell & Bioscience (2024)
-
Recurrent de novo pathogenic variant of WASF1 in a Japanese patient with neurodevelopmental disorder with absent language and variable seizures
Human Genome Variation (2021)
