
Abstract
A 1-month-old Japanese infant with cardiac rhabdomyoma was diagnosed with TSC2/PKD1 contiguous gene syndrome by targeted panel sequencing with subsequent quantitative polymerase chain reaction that revealed gross monoallelic deletion, including parts of two genes: exons 19–42 of TSC2 and exons 2–46 of PKD1. Early molecular diagnosis can help to detect bilateral renal cyst formation and multidisciplinary follow-up of this multisystem disease.
Similar content being viewed by others

Tuberous sclerosis complex (TSC, OMIM #191100 and #613254) is an autosomal dominant multiple system disorder characterized by hamartomatous growth abnormalities in multiple organ systems, including the brain, skin, heart, lungs, and kidneys1. TSC has a broad phenotypic spectrum, including seizures, mental retardation, renal dysfunction, and dermatological abnormalities and tumors, and the clinical manifestations of TSC vary individually. TSC is caused by pathogenic variants in either one of the two disease-causing genes, TSC1 (OMIM #605284, 9p34) or TSC2 (OMIM #191092, 16p13.3), encoding hamartin and tuberin, respectively2. Pathogenic variants in TSC2 occur 4–5 times more often and are associated with a more severe phenotype than those in TSC11. The TSC2 gene lies immediately adjacent to PKD1 (OMIM #601313), which encodes polycystin-1 and is the major gene causing autosomal dominant polycystic kidney disease (ADPKD, MIM#173900). ADPKD is characterized by progressive bilateral renal cysts and is sometimes complicated by liver cysts and intracranial aneurysms. Both genes are in tail-to-tail orientation. Large deletions disrupting both TSC2 and PKD1 result in TSC2/PKD1 contiguous gene syndrome (PKDTS, MIM#600273). This disease was first reported by Brook–Carter et al. in 1994 with a variety of phenotypes dominated by severe, very early-onset PKD and occurs in ~2–5% of TSC patients, resulting in significant renal insufficiency in teenage years3,4,5,6,7,8.
We present herein the case of a male infant with TSC in whom multiple cardiac rhabdomyomas were detected at 27 weeks of gestation on ultrasound. Retinal hamartoma, multiple subependymal nodules, cortical tubers, and bilateral multiple renal cysts were detected after birth. Targeted panel sequencing (TPS) identified the gross germline deletion involving parts of the TSC2 and PKD1 genes, which has never been reported before.
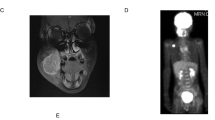
The male infant was born as the first child of healthy, nonconsanguineous Japanese parents with an unremarkable family history. At 27 weeks of gestation, cardiac tumors were detected on fetal ultrasonography, and a massive tumor was seen protruding from the ventricular septum to the left ventricle with multiple small tumors on both ventricle walls (Fig. 1a). Fetal ultrasound and magnetic resonance imaging also showed a 2 cm diameter tumor with a mixture of high and low echo areas and increased and decreased signal intensity areas, respectively, in the right kidney. He was delivered at 37 weeks and 5 days of gestation by vaginal delivery. At birth, the weight was 3468 × g (+2.2 SD), length was 53 cm (+2.9 SD), and head circumference was 36 cm (+2.4 SD) with no marked physical presentation. Retinal hamartoma was observed as an ocular lesion near the macula of the right eye. On neonatal ultrasonography, no clear renal cysts were observed, although a tumor was detected in the lower pole of the right kidney. Because the obstruction of the left ventricular outlet by the tumor gradually worsened, the cardiac tumor underwent partial surgical resection when he was 29 days old. Pathological findings for the tumor were compatible with rhabdomyoma, and residual tumors regressed spontaneously. Magnetic resonance imaging showed multiple bilateral renal cysts as well as multiple subependymal nodules and cortical tubers (Fig. 1b–d). Although seizures were not evident clinically, electroencephalography at 5 months old showed paroxysmal focal spikes in the right frontal region, resulting in the initiation of valproate treatment. Based on these findings, TSC was clinically suspected, whereas PKD was not suspected at that time9. The diagnosis of PKDTS was genetically confirmed by applying TPS for a combined screening of causative alterations in candidate genes as described below at 2 months old. As of the time of writing, at 3 years old, he was healthy with normal renal function, no progression of the ocular lesion, and normal neurological development without epileptic attacks.

a Fetal echocardiography showing rhabdomyoma (white arrows), Ao aorta, LA left atrium, LV left ventricle. b Magnetic resonance imaging (MRI, T2-weighted image) of abdomen at 3 months old showing enlarged kidneys with multiple cysts. c MRI (T1-weighted image) of brain at 55 days old showing multiple subependymal nodules along the lateral ventricle walls (white arrows). d MRI (T2-weighted image) of brain at 3 years old showing multiple subcortical high signal lesions indicating cortical tubes in cerebral hemisphere, predominantly in frontal lobe (white arrows).
The ethics committee at Tokushima University approved this study. After written informed consent was obtained from the parents of the patient, genetic analysis was performed using genomic DNA extracted from whole blood obtained from the patient at 2 months old. We conducted TPS for the exons of 4813 disease-related genes using the Trusight One Sequencing Panel (Illumina, San Diego, CA) and a MiSeq sequencer (Illumina), followed by analysis using our pipeline for next-generation sequencing (NGS) data, as described previously10,11,12. GRCh37/hg19 (build 37 of the Genome Reference Consortium human genome) was used as the human reference genome sequence. Detection of copy number variations (CNVs) using TPS data with resolution ranging from a single exon to several exons depending on exon size was performed as described previously11. These analyses showed no disease-causing SNVs or small insertions/deletions (indels) but detected a gross monoallelic deletion at least 47.6 kb long, including parts of two genes: exons 19–42 of TSC2 (NM_000548.5) and exons 2–46 of PKD1 (NM_001009944.3), located from positions 2,121,775–2,169,389 of chromosome 16 (16p13.3), suggesting TSC2/PKD1 contiguous gene syndrome as a molecular diagnosis (Fig. 2a). However, the data from the probe for exon 1 of PKD1 for TPS were not available, possibly due to a relatively high GC content. To validate a deletion and determine deletion breakpoints, we performed a SYBR green-based real-time quantitative polymerase chain reaction (qPCR) as described elsewhere (Fig. 2a, Supplementary table S1)13. Monoallelic deletions of parts of TSC2 and PKD1 were validated, and the breakpoints appeared to be located between exons 18 and 19 of TSC2 and between exons 1 and 2 of PKD1 (Fig. 2b), suggesting that the maximum size of the deleted region is ~64 kb. The deletion was not confirmed as de novo because of the unavailability of parental DNA.

a An eXome-Hidden Markov Model v1.0 (XHMM, https://atgu.mgh.harvard.edu/xhmm/) analysis using TPS data detected a gross monoallelic deletion at least 47.6kb long (black closed arrow), including parts of two genes: exons 19–42 of TSC2 and exons 2–46 of PKD1, located from positions 2,121,775–2,169,389 of chromosome 16 (red solid bar). The x-axis denotes the physical position, and the y-axis indicates the Z-score of principal component analysis (PCA)-normalized read depth. Purple circles connected by red lines show values for the individual subjected to TPS in this study. Gray dots with gray connected lines represent the results of normalized read depth obtained from in-house control data (N=126). Blue vertical bars in each gene represent exons. Because the data from the probe for exon 1 of PKD1 for TPS were not available, we could not determine the break point around exon 1 of PKD1 by TPS, although the break point within TSC2 appeared to be located between exons 18 and 19. To validate a deletion and determine deletion breakpoints, we performed SYBR green-based qPCR using primer sets targeted to six sites around two genes (black vertical bars) and the control site, SHGC16885 (Supplementary table S1). b Results of qPCR to determine a relative genomic copy number using genomic DNA from the proband in the present study (left) and the healthy male control (right). Based on the relative copy numbers determined by qPCR, monoallelic deletions of parts of TSC2 and PKD1 were validated, and the breakpoints appeared to be located between exons 18 and 19 of TSC2 and between exons 1 and 2 of PKD1, suggesting that the maximum size of the deleted region is ~64kb (purple closed arrow).
According to the Human Gene Mutation Database professional 2020.1 (HGMD, https://portal.biobase-international.com/hgmd/pro/all.php), 1227 damaging variants have been reported in TSC2. Of these, 176 (14.3%) were gross deletions, and 32 (2.6%) involved simultaneous deletion of the adjacent PKD1 gene, although the number of variants does not directly indicate the prevalence of PKDTS. In addition, heterozygous pathogenic variants in both TSC1 and TSC2 are responsible for TSC. Because only TSC was clinically suspected in the present case at birth, we applied TPS based on next-generation sequencing (NGS) technology to simultaneously investigate SNVs, indels and CNVs in most exons of candidate disease-causing genes, TSC1 and TSC2, and their adjacent genes. Our findings demonstrated the utility of NGS for improving the molecular diagnosis of TSC and PKDTS in a timely and cost-effective manner. Previously, both multiplex ligation-dependent probe amplification (MLPA) and array comparative genomic hybridization (array-CGH) were shown to be useful not only for the diagnosis of PKDTS but also for elucidation of its molecular mechanism14,15. However, it is also known that a prompt diagnosis of PKDTS can be complicated by the phenotypic heterogeneity of PKD and the absence of a clear phenotype-genotype correlation15. Under conditions where there is no strong clinical suspicion of PKDTS, NGS-based technology can be used to simultaneously evaluate multiple genetic alterations causing TSC.
Although ~25–32% of TSC cases exhibit some degree of renal cyst formation, most of which occurs in the second decade of life4,5, PKDTS seems typically associated with severe juvenile polycystic kidney disease3,6,7,8. Sampson et al. reported that among 27 unrelated patients with TSC and renal cystic disease, cystic disease was more severe, with early renal insufficiency, in 17 patients with PKDTS showing constitutional deletions7. Patients with PKDTS should therefore be diagnosed as early as possible to predict the increased risk of ADPKD-related complications, including cystic kidney disease as well as early end-stage renal disease6,7,15. In addition, most patients with TSC also develop neurological and neuropsychiatric disorders, with up to 90% developing epilepsy and up to 50% developing autism, suggesting that treating infants with TSC and epilepsy early with antiepileptic medications or surgery may result in better neurological outcomes16. In summary, early molecular diagnosis of PKDTS may be crucial for providing appropriate disease-related surveillance and therapeutic options in patients, as well as appropriate genetic counseling for the family.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at, https://doi.org/10.6084/m9.figshare.hgv.2870, https://doi.org/10.6084/m9.figshare.hgv.2873.
References
De Waele, L. Lagae, L. & Mekahli, D. Tuberous sclerosis complex: the past and the future. Pediatr. Nephrol. 30, 1771–1780 (2015).
Lam, H. C., Nijmeh, J. S. & Henske, E. P. New developments in the genetics and pathogenesis of tumours in tuberous sclerosis complex. J. Pathol. 241, 219–225 (2017).
Brook-Carter, P. T. et al. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease—a contiguous gene syndrome. Nat. Genet. 8, 328–332 (1994).
Cook, J. A., Mueller, R. F. & Sampson, J. A cross sectional study of renal involvement in tuberous sclerosis. J. Med. Genet. 33, 480–484 (1996).
Dabora, S. L. et al. Mutation analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared to TSC1, disease in multiple organs. Am. J. Hum. Genet. 68, 64–80 (2001).
Longa, L. et al. A large TSC2 and PKD1 deletion is associated with renal and extrarenal signs of autosomal dominant polycystic kidney disease. Nephrol. Dial. Transpl. 12, 1900–1907 (1997).
Sampson, J. R. et al. Renal cystic disease in tuberous sclerosis: role of the polycystic kidney disease 1 gene. Am. J. Hum. Genet. 61, 843–851 (1997).
Torra, R. et al. Facilitated diagnosis of the contiguous gene syndrome: tuberous sclerosis and polycystic kidneys by means of haplotype studies. Am. J. Kidney Dis. 31, 1038–1043 (1998).
Northrup, H. & Krueger, D. A. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 international tuberous sclerosis complex consensus conference. Pediatr. Neurol. 49, 243–254 (2013).
Okamoto, N., Naruto, T., Kohmoto, T., Komori, T. & Imoto, I. A novel PTCH1 mutation in a patient with Gorlin syndrome. Hum. Genome Var. 1, 14022 (2014).
Watanabe, M. et al. A novel missense mutation of COL5A2 in a patient with Ehlers–Danlos syndrome. Hum. Genome Var. 3, 16030 (2016).
Watanabe, M. et al. Detection of 1p36 deletion by clinical exome-first diagnostic approach. Hum. Genome Var. 3, 16006 (2016).
Gao, W. et al. DGCR6 at the proximal part of the DiGeorge critical region is involved in conotruncal heart defects. Hum. Genome Var. 2, 15004 (2015).
Oyazato, Y. et al. Molecular analysis of TSC2/PKD1 contiguous gene deletion syndrome. Kobe J. Med. Sci. 57, E1–E10 (2011).
Reyna-Fabian, M. E. et al. TSC2/PKD1 contiguous gene syndrome, with emphasis on a case with an atypical mild polycystic kidney phenotype and a novel genetic variant. Nefrologia 40, 91–98 (2020).
Davis, P. E. et al. Presentation and diagnosis of tuberous sclerosis complex in infants. Pediatrics 140, e20164040 (2017).
Acknowledgements
We wish to thank the patient and his family for their participation in this study. This work was supported by JSPS KAKENHI grant number 18H02894 to I.I. from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Osumi, K., Suga, K., Ono, A. et al. Molecular diagnosis of an infant with TSC2/PKD1 contiguous gene syndrome. Hum Genome Var 7, 21 (2020). https://doi.org/10.1038/s41439-020-0108-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-020-0108-0
