
Abstract
USP9X variants have been reported in patients with X-linked intellectual disability. Here, we report two female patients with intellectual disability and pigment abnormalities along Blaschko lines. Targeted resequencing identified two novel heterozygous variants, c.4068_4072del (p. (Leu1357Tyrfs*12)) and c.1201C>T (p. (Arg401*)), in USP9X. Our findings provide further evidence that USP9X variants cause intellectual disability.
Similar content being viewed by others

Intellectual disability (ID) is characterized by significant limitations in both intellectual functioning and adaptive behavior, with onset before the age of 18 years, and is commonly defined by an IQ score of <701,2. ID occurs in ~1–3% of the population worldwide1. To date, almost 100 causative genes have been reported for X-linked ID (XLID)3.
Variants in USP9X, which is located at Xp11.4, have been suggested to cause various types of cancer, female-restricted X-linked syndromic mental retardation-99 (MIM 300968), and X-linked recessive mental retardation-99 (MIM 300919). USP9X encodes ubiquitin-specific protease 9×, which is highly expressed in the mouse brain and plays important roles in nervous system development, stabilization of myeloid leukemia cell differentiation protein (MCL1) in human follicular lymphomas and diffuse large B-cell lymphomas, and tumor cell survival4,5,6.
In this study, we identified novel USP9X variants in two female patients with XLID by using targeted resequencing.
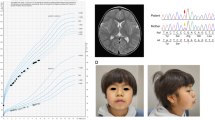
Patient 1 is a 4-year-old girl who is the second child of healthy and nonconsanguineous parents. She was delivered at 37 weeks of gestation with a birth weight of 2278 g (−1.1 SD), a length of 47 cm (−0.1 SD), and an occipital frontal circumference of 30.5 cm (−1.5 SD). She was able to walk unsupported by 23 months. She began speaking in recognizable words at 2 years of age, at which point she also began to show signs of moderate-to-severe ID. The patient was referred to Kanagawa Children’s Medical Center because of developmental delay at 2 years of age. At the time of referral, she was 82.7-cm tall (−1.0 SD), weighed 13.4 kg (1.6 SD), and had an occipital frontal circumference of 46.4 cm (−0.7 SD). She had dysmorphic features consisting of upswept and curly hair, facial asymmetry, prominent forehead, bitemporal narrowing, short palpebral fissures, prominent nose with flared ala nasi, smooth philtrum, thin upper lip, full cheeks, dysplastic ears, tapering fingers, and Blaschko lines (Fig. 1a, b). She spoke several individual words and exhibited autistic behavior, including repetitive and stereotyped movements. At 3 years and 11 months of age, she developed obesity. She was 98-cm (0 SD) tall and weighed 18.6 kg (1.9 SD), and her body mass index was 19.4, which is >97th percentile for age. Her karyotype is 46,XX.

a Patient 1 at 4 years of age. The patient exhibited upswept and curly hair, facial asymmetry, prominent forehead, bitemporal narrowing, short palpebral fissures, prominent nose with flared ala nasi, smooth philtrum, thin upper lip, full cheeks, and dysplastic ears. b Patient 1 at 4 years of age. The patient had tapered fingers. c Patient 2 at 18 months of age. Pigment changes along Blaschko lines bilaterally on the upper arms (d) and neck
Patient 2 is the first child of a 40-year-old primigravid mother and a nonconsanguineous 41-year-old father. Pregnancy was achieved by artificial insemination by using the husband’s sperm, and labor and delivery were uneventful. Owing to the advanced maternal age, prenatal cytogenetic analysis by using G-banding was performed on amniocentesis, and the results were normal. The maternal grandfather’s sister had a history of retinitis pigmentosa. The proposita was born at 41 weeks and 1 day of gestation. The newborn had a birth weight of 2968 g (−0.5 SD), a length of 48.3 cm (−0.9 SD), and a head circumference of 34.2 cm (0.4 SD). Apgar scores were 8 at 1 min and 9 at 5 min. During the early neonatal period, bilateral cryptotia, congenital hearing impairment as detected by auditory brain-stem response (rt-70 dB, lt-50 dB), and right muscular torticollis were noted. Bilateral clasped thumbs were identified at 2 months of age and had improved by 11 months of age. She was first evaluated by Kawasaki Medical School Hospital at 9 months of age because of developmental delay. The main clinical manifestations included epicanthus, telecanthus, short columella, depressed nasal tip, bilateral low-set and posteriorly rotated ears, overfolded helices, bifid uvula, umbilical hernia, and bilateral overlapping toes (T2–3, T4–5). At the time of evaluation, she was 73.4 cm (1.4 SD) in height, weighed 8465 g (0.3 SD), and had a head circumference of 44.8 cm (0.8 SD). Her muscle tone was normal. Pigment changes along Blaschko lines appeared bilaterally on the upper arms at 12 months and on the neck at 18 months (Fig. 1c, d). She suffered from recurrent otitis media with effusion after 12 months of age. She was able to lift her head at 4 months of age, sat alone at 8 months, babbled at 12 months, and walked unaided at 17 months. Her developmental quotient was 71 at 17 months. At 18 months, her height was 80.8 cm (0.5 SD), and her weight was 9.9 kg (0 SD). Ophthalmological evaluations revealed astigmatism, hyperopia, and normal fundi. Ultrasonographic examination of the heart and abdomen showed normal findings. Other normal laboratory tests included thyroid function (free T4, free T3), somatomedin-C, serum chemistries, immunoglobulins, complete blood counts, blood gas analysis, and urinalysis.
Clinical information was obtained after obtaining written informed consent from the patients’ families. The institutional review board of Kanagawa Children’s Medical Center approved this study. Genomic DNA was extracted from both patients’ peripheral blood by using the QIAcube (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. Targeted resequencing was performed for the two affected patients. Genomic DNA was captured by the TruSight One Sequencing Panel (Illumina, Inc., San Diego, CA, USA) and was sequenced on a MiSeq platform (Illumina) with 151-bp paired-end reads, as previously described7. The candidate variant was confirmed by Sanger sequencing.
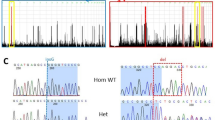
Targeted resequencing identified two heterozygous variants in USP9X (NM_001039590.2): c.4068_4072del (p.(Leu1357Tyrfs*12)) in patient 1 and c.1201C>T (p.(Arg401*)) in patient 2. These variants were not present in the NHLBI-Exome Sequencing Project 6500, the 1000 Genomes Project, dbSNP138, the Human Genetic Variation Database or in our in-house Japanese exome database. Sanger sequencing confirmed that these variants had occurred de novo (Fig. 2).

Novel heterozygous variants identified in two female patients. USP9X is predicted to contain a ubiquitin-specific protease domain, as determined by SMART (http://smart.embl-heidelberg.de/). Electropherogram for each patient and her parents
Here, we report two female patients with heterozygous variants in USP9X who exhibited ID and pigment abnormalities along Blaschko lines (Supplementary Table S1). These two USP9X variants are truncating variants, resulting in a premature stop codon (Fig. 2). Reijnders et al. reported de novo loss-of-function variants of USP9X in 17 female patients8. Au et al. also reported de novo pericentric inversion resulting in a 0.326-Mb deletion of the USP9X 5′ UTR and a de novo truncating variant in two female patients9. Homan et al. reported two maternally inherited missense variants and a truncating variant of USP9X in three male patients10. This truncating variant is located in the last exon, and as such, the mRNA presumably escapes nonsense-mediated decay. Thus, USP9X alterations in female-restricted X-linked syndromic mental retardation-99 may confer loss-of-function effects. On the other hand, USP9X alterations in X-linked recessive mental retardation-99 may confer hypomorphic or a milder form of loss-of-function effects. The clinical features of our two patients were compared with previously reported female patients with variants in USP9X (Supplementary Table S1). All patients with USP9X variants exhibited ID. Some patients with ID also showed pigment abnormalities along Blaschko lines, including our patients. Dental abnormalities, asymmetric hypomastia, heart defects, urogenital abnormalities, scoliosis, postaxial polydactyly, seizures, hypotonia, and recurrent respiratory tract infections were observed in some of the previously reported patients but not in our patients. Further analysis is required to determine the phenotype–genotype correlation.
Endogenous USP9X localizes to the primary cilium; however, this localization was significantly decreased when USP9X expression was knocked down in fibroblasts by using siRNA8. Female heterozygous knockout mice (Nes-Usp9x−/X) were normal at birth and survived to adulthood6. A small reduction in the hippocampal area was observed in adult female knockout mice (Emx1-Usp9x−/X)6. However, no reduction in the hippocampal area was observed in our patients. In contrast, male hemizygous knockout mice (Nes-Usp9x −/Y) died within 24 h of birth6.
In conclusion, we identified heterozygous USP9X variants in two female patients. Our report provides further evidence that USP9X variants are associated with XLID.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.2621, https://doi.org/10.6084/m9.figshare.hgv.2624.
References
Iwase, S. et al. Epigenetic etiology of intellectual disability. J. Neurosci. 37, 10773–10782 (2017).
Vissers, L. E., Gilissen, C. & Veltman, J. A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 17, 9–18 (2016).
Neri, G., Schwartz, C. E., Lubs, H. A. & Stevenson, R.E. X-linked intellectual disability update 2017. Am. J. Med. Genet. A 176, 1375–1388 (2018).
Zhang, C. et al. USP9X destabilizes pVHL and promotes cell proliferation. Oncotarget 7, 60519–60534 (2016).
Jo, Y. S., Kim, M. S., Yoo, N. J. & Lee, S. H. USP9X, a putative tumor suppressor gene, exhibits frameshift mutations in colorectal cancers. Pathol. Oncol. Res. 23, 219–220 (2017).
Stegeman, S. et al. Loss of Usp9x disrupts cortical architecture, hippocampal development and TGFbeta-mediated axonogenesis. PLoS ONE 8, e68287 (2013).
Sato, Y. et al. Novel COL4A1 mutation in a fetus with early prenatal onset of schizencephaly. Hum. Genome Var. 5, 4 (2018).
Reijnders, M. R. et al. De novo loss-of-function mutations in USP9X cause a female-specific recognizable syndrome with developmental delay and congenital malformations. Am. J. Hum. Genet. 98, 373–381 (2016).
Au, P. Y. B. et al. Two females with mutations in USP9X highlight the variable expressivity of the intellectual disability syndrome. Eur. J. Med Genet. 60, 359–364 (2017).
Homan, C. C. et al. Mutations in USP9X are associated with X-linked intellectual disability and disrupt neuronal cell migration and growth. Am. J. Hum. Genet. 94, 470–478 (2014).
Acknowledgements
We thank the patients and their families for participating in this work. We also thank Noriaki Harada and Shizuka Shiiya for technical assistance. This research was supported by a Grant-in-Aid from the Ministry of Health, Labour, and Welfare, Japan and Japan Agency for Medical Research and Development (AMED) 18kk02050h003, Initiative on Rare and Undiagnosed Diseases 18ek0109301, and JSPS KAKENHI 17K10069 (K.K.); and the Yokohama Foundation for Advancement of Medical Science (Y.T.). We thank Emily Crow, Ph.D., from Edanz Group (www.edanzediting.com/ac) for editing a draft of this paper.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tsurusaki, Y., Kuroda, Y., Yamanouchi, Y. et al. Novel USP9X variants in two patients with X-linked intellectual disability. Hum Genome Var 6, 49 (2019). https://doi.org/10.1038/s41439-019-0081-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-019-0081-7
This article is cited by
-
Characteristic craniofacial defects associated with a novel USP9X truncation mutation
Human Genome Variation (2024)
-
Missense variant contribution to USP9X-female syndrome
npj Genomic Medicine (2020)
