
Abstract
Raphanobrassica (RrRrCrCr, 2n = 4x = 36), which is generated by distant hybridization between the maternal parent Raphanus sativus (RsRs, 2n = 2x = 18) and the paternal parent Brassica oleracea (C°C°, 2n = 2x = 18), displays intermediate silique phenotypes compared to diploid progenitors. However, the hybrid shares much more similarities in silique phenotypes with those of B. oleracea than those of R. sativus. Strikingly, the silique of Raphanobrassica is obviously split into two parts. To investigate the gene expression patterns behind these phenomena, transcriptome analysis was performed on the upper, middle, and lower sections of pods (RCsiu, RCsim, and RCsil), seeds in the upper and lower sections of siliques (RCseu and RCsel) from Raphanobrassica, whole pods (Rsi and Csi) and all seeds in the siliques (Rse and Cse) from R. sativus and B. oleracea. Transcriptome shock was observed in all five aforementioned tissues of Raphanobrassica. Genome-wide unbalanced biased expression and expression level dominance were also discovered, and both of them were toward B. oleracea in Raphanobrassica, which is consistent with the observed phenotypes. The present results reveal the global gene expression patterns of different sections of siliques of Raphanobrassica, pods, and seeds of B. oleracea and R. sativus, unraveling the tight correlation between global gene expression patterns and phenotypes of the hybrid and its parents.
Similar content being viewed by others


Introduction
Hybridization provides a way of interspecific or intergeneric genome transfer for incorporating preferable traits from the parents to the progeny. Allopolyploid hybrids generated from interspecific or intergeneric hybridization have provided abundant genetic resources for molecular biology research and crop breeding. Many crops, such as wheat1, cotton2, rapeseed3, and Chinese cabbage4, are derived from hybridization between different species. However, polyploidization might induce several important changes at the genetic, gene expression, and epigenetic levels5,6,7,8,9,10. In recent decades, many efforts have been made to produce synthetic allopolyploid species, such as Arabidopsis11,12, Brassica13,14, Oryza15, Nicotiana16, Triticum17 and Gossypium18. Among them, Brassica species are one of the model systems to study crop allopolyploidization19,20. For example, miRNA expression patterns were explored between B. napus, B. rapa, and their F1 hybrids21. However, the genomic instability and the difficulty of the production of distant hybrids impede a better understanding of global expression patterns and/or regulatory mechanisms of gene expression during hybridization.
Allopolyploids often show genome-wide expression level dominance and/or homoeolog expression bias, namely, the hybrid is more similar and/or biases to one parent than the other22. Such expression dominance and/or bias phenomena have been reported in species such as B. napus23, P. hesperium24, G. hirsutum25,26, and T. turgidum27. One representative example has been recently reported in newly resynthesized B. napus allopolyploids, in which genome-wide expression bias and dominance are both toward the A-genome13. It has also been revealed that a high proportion of genes in Triticum turgidum, which has heterosis, exhibit parental expression level dominance in the hybrid between T. turgidum (AABB) and Aegilops tauschii (DD)17. Although plant polyploidy has potential advantages in growth vigor (heterosis), allopolyploidization usually induces “genomic shock” due to imbalanced and antagonistic gene expression in polyploids28.
Although recent research on polyploid hybrid plants has exploded, the majority of the previous reports are based on interspecific and/or intrageneric species. Some studies have also focused on intergeneric hybridizations within the Brassicaceae family in the last few years29,30. Nevertheless, the global gene expression patterns of different intergeneric hybrids within the Brassicaceae family have scarcely been reported. In our group, a synthetic intergeneric hybrid Raphanobrassica (RrRrCrCr, 2n = 36) was constructed by hybridization between R. sativus and B. alboglabra Bailey, which has high fertility and resistance to clubroot disease and beet cyst nematode effectiveness31,32,33. The newly synthesized hybrid provides a perfect model to study the plant polyploidy generated by crossing distinct important vegetables. In this study, we aimed to explore the global gene expression patterns of Raphanobrassica and its parents, determine the relationships between the gene expression and the silique phenotypes in which the hybrid shows intermediate traits compared to its diploid progenitors and shares many more similarities with B. oleracea. RNA sequencing was performed, and transcriptome shock was observed in RCsiu, RCsim, RCsil, RCseu, and RCsel of Raphanobrassica. The global gene expression patterns of the five tissues of the hybrid were much more similar to those of B. oleracea than those of R. sativus. The five tissues also showed genome-wide unbalanced biased expression and expression level dominance toward B. oleracea.
Results
The silique phenotypes of Raphanobrassica and its diploid progenitors
The intergeneric allotetraploid Raphanobrassica (RrRrCrCr) was generated by crossing diploid R. sativus (RsRs, maternal parent) and diploid B. oleracea var. alboglabra (C°C°, paternal parent) following fertility selection for ten generations (Fig. 1a). For convenience, we use the species names and their representative genomes interchangeably in the following text, i.e., RsRs for R. sativus, C°C° for B. oleracea, and RrRrCrCr for Raphanobrassica. Siliques of the hybrid exhibit intermediate phenotypes compared to those of the diploid progenitors (Fig. 1b–e and Fig. S1). Specifically, the silique of the hybrid is obviously split into two parts joined by the valve shoulder (RCsim). The upper section of pods (RCsiu) displays more similarities to those of R. sativus, and neither of them possesses the seed-bearing septum (Fig. 1b, c). Instead, seeds in RCsiu and Rsi are enclosed by sponge-like membranes. In contrast, RCsil shares more similarities with the silique of C°C° (Csi), and both harbor a seed-bearing septum (Fig. 1b, c). However, the whole silique length of RrRrCrCr (4.65 ± 0.10 cm) is between those of the progenitor RsRs (3.33 ± 0.14 cm) and C°C° (5.37 ± 0.12 cm), but it is much more similar to that of C°C°. A similar phenomenon was found in the phenotype of silique width, with RCsiu (4.83 ± 0.17 mm) and RCsil (4.67 ± 0.21 mm) sharing much more similarities with C°C° (3.83 ± 0.17 mm) than RsRs (10.00 ± 0.26 mm) (Fig. 1d and Fig. S1). As expected, the number of seeds per silique of RrRrCrCr (10.33 ± 1.21) was between those of RsRs (4.17 ± 1.17) and C°C° (18.67 ± 1.03), with more seeds in RCsil (7.67 ± 0.82) than in RCsiu (2.67 ± 0.82) (Fig. 1e). In addition, the color of mature seeds, seed area, and thousand seed weight (TSW) of the hybrid were also between those of the two parents but were much more similar to those of C°C° (Fig. S1). Of note, few differences in the phenotypes of seed area, TSW, and color of mature seeds between RCseu and RCsel were found.

a The schematic diagram for the generation of allotetraploid Raphanobrassica. b Comparisons of the whole silique, pod, and seed phenotypes among the hybrid and its diploid parents at 35 DAF. The black arrow indicates a sponge-like membrane enclosing RsRs seeds. Scale bars, 1 cm. c Magnified images of those from (b) in white rectangular boxes. Arrows indicate the dehiscence zones of Csi and RCsil. d Silique length (without the tip) of RsRs, C°C°, RrRrCrCr, upper and lower parts of the RrRrCrCr silique at 35 DAF. Error bars, standard deviation. eThe number of seeds per silique of RsRs, C°C°, RrRrCrCr, upper and lower parts of RrRrCrCr silique. f A 3:3 synteny pattern between RsRs and C°C° genomes. g Dot plot of pairwise synteny between RsRs and C°C° genomes. R1-R9 indicated the nine longest scaffolds of the RsRs genome. C1-C9 indicated the nine chromosomes of C°Co genome
In brief, silique phenotypes of silique structure, silique length, silique width, seed area, the color of mature seeds, and TSW of the hybrid are between those of its parents but very similar to those of C°C°. Conclusively, phenotypes of the whole silique of Raphanobrassica are much more similar to those of B. oleracea than those of R. sativus, even though the developing seeds of the hybrid also share slight similarities with those of R. sativus (Fig. 1b), suggesting that global gene expression pattern of Raphanobrassica silique is much more similar to its paternal parent than that of its maternal parent.
Global mRNA transcriptome analysis of different sections of silique
To investigate the gene expression patterns behind these interesting phenomena, we first performed synteny analysis between the paternal (C°C°, assembly BOL) and maternal (RsRs, assembly Rs1.0) genomes. It was revealed that the synteny depth between the paternal and maternal genomes is a 3:3 pattern (Fig. 1f), and the genome-wide synteny between these two genomes is of high level (Fig. 1g), indicating that many genes of the two genomes are highly homologous. Subsequently, to unravel the global gene expression patterns, RNA-seq of RCsiu, RCsim, RCsil, RCseu, RCsel, Rsi, Rse, Csi, and Cse was performed. All of the generated sequencing reads were processed and mapped to the RsRs genome (assembly Rs1.0), C°C° genome (assembly BOL), and integrated genomes of RsRs and C°C° (Table S1). Except for Csi3, we obtained approximately 1.4 × 109 clean reads (5.4 × 107 on average), approximately 1.2 × 109 of which (83.4%) were mapped to the corresponding genomes. Most of the mapped reads were uniquely mapped reads (94.2%), and the vast majority of them were mapped to exons (Table S1). Pearson correlation coefficients (PCCs) between these samples ranged from 0.801–0.996, which indicates the high-quality of the biological replicates (Fig. S2). Twelve genes were randomly selected for further RT–qPCR analysis, and the results showed the same expression patterns as the mRNA-seq data (Fig. S3).
There were indeed some differences in global gene expression patterns among RCsiu, RCsim, and RCsil, but few were found between those of RCseu and RCsel (Fig. 2a–c). Thus, for convenience, only the transcriptome of RCseu was used for further homoeolog expression bias and expression level dominance analyses in the following text. Moreover, the results also revealed that the global gene expression level distribution of RrRrCrCr was more similar to that of C°C° than that of RsRs in all five silique sections (Fig. 2a and Fig. S4).

a Heat map showing the expression levels of 20,000 randomly selected genes from Rsi, Rse, Csi, Cse, RCsiu, RCsim, RCsil, RCseu, and RCsel. b Numbers of expressed genes and DEGs of the nine tissues. Total numbers of expressed genes of different tissues are indicated by the black numbers, and the blue and red numbers along the solid black lines reveal the down- and upregulated genes in the five tissues of RrRrCrCr relative to those in the progenitors, respectively. In addition, the red and green numbers in the brackets indicate the genes coming from the RrRr and CrCr subgenomes, respectively. c Total DEGs identified in pairwise comparison among the three sections of pods and between the seeds in the two sections of siliques of the hybrid. d Functional categorization of DEGs in GO terms among the nine tissues. e Expression levels of 20 randomly selected photosynthesis-related DEGs in RrRrCrCr relative to the diploid progenitors. RrRr and CrCr indicated the two subgenomes of the hybrid
In total, we identified 59,018 (41,140 known genes and 17,878 novel gene loci) genes as expressed (empirical cutoff value: FPKM ≥1) in at least one of twenty-six samples (except for Csi3). Specifically, a total of 28,517, 14,378, 30,084, 17,574, 21,184, 23,899, 23,873, 22,504 and 22,811 expressed genes were identified in Rsi, Csi, Rse, Cse, RCsiu, RCsim, RCsil, RCseu and RCsel, respectively (Fig. 2b). The three sections of pods and the seeds in the two sections of RrRrCrCr siliques have intermediate numbers of expressed genes compared to those of the two progenitors, which is consistent with the intermediate phenotypes of silique of the hybrid (Fig. 2b). Dramatically, most of the differentially expressed genes (DEGs) in RCsiu, RCseu, and RCsel were downregulated relative to those in the diploid progenitors. However, there were more upregulated DEGs than downregulated DEGs in RCsim and RCsil when compared to Csi (Fig. 2b). Different sets of DEGs were also classified by GO terms. Notably, the upregulated and downregulated DEGs in seeds of RrRrCrCr relative to the RsRs genome and the C°C° genome, respectively, were enriched for photosynthesis-related GO terms (Fig. 2d). However, the expression levels of the 20 randomly selected photosynthesis-related DEGs in Raphanobrassica were comparable between the subgenomes of RrRr and CrCr (Fig. 2e).
Together, the sharp alteration in the global gene expression patterns of pods and seeds of RrRrCrCr relative to those of the parental genomes indicates a “transcriptome shock” in the hybrid genome. The intermediate numbers of expressed genes of pods and seeds of the hybrid between those of the progenitors are also consistent with the intermediate phenotypes of pods and seeds of the hybrid. However, the genomic regulation mechanisms underlying these phenomena remain unclear.
Analysis of expression profile differences between the upper and lower sections of silique of RrRrCrCr
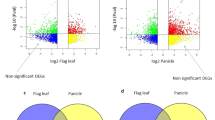
Although there are few differences in the expression profile between RCseu and RCsel, we want to know what the specific differences are, as well as those between RCsiu and RCsil. KEGG terms of upregulated DEGs of RCseu relative to RCsel were significantly enriched for ‘plant hormone signal transduction’ and ‘MAPK signaling pathway-plant’ (Fig. 3a). Detailed analysis revealed that two of these DEGs (three unique genes in total) negatively regulate ABA signaling, and one positively regulates cytokinin signaling. Suppression of AHG3 (gene ID: 106295669), a negative regulator of ABA signaling, could significantly accelerate fruit ripening in tomato34. However, KEGG analysis of downregulated DEGs of RCseu relative to RCsel was significantly enriched for ‘Protein processing in endoplasmic reticulum’ (Fig. 3b). Interestingly, seven homoeolog gene pairs were detected within the downregulated DEGs of RCseu relative to RCsel, and three pairs (gene ID: 108812972–106312198, 108817415–106299879, and 108857941–106295243) were identified as molecular chaperones that play critical roles in ER stress35. A previous study revealed that these molecular chaperones might be required for desiccation tolerance in Arabidopsis36. These results indicate that the ripening of RCseu might lag behind that of RCsel, which is in line with the phenotype in which the color of RCsel is deeper than that of RCseu (Fig. 1b). Furthermore, Venn diagram analysis revealed a high proportion of coexpressed genes between RCseu and RCsel (20,845, 95.6% in RCseu and 95.9% in RCsel), and the two tissues shared more coexpressed genes with Rse than Cse (Fig. 3c). In addition, the difference in the expression level of specifically expressed genes in RCseu and RCsel was minimal, and the top eight highly specifically expressed genes in RCsel were identified as novel gene loci (Fig. S5). Together, these results indicate that there are subtle, but nearly no, differences in expression profiles between RCseu and RCsel, which is in line with the phenotypes.

a KEGG enrichment analysis of upregulated DEGs of RCseu relative to those of RCsel. b KEGG enrichment analysis of downregulated DEGs of RCseu relative to those of RCsel. c Venn diagram analysis of expressed genes in Rse, Cse, RCseu, and RCsel. d KEGG terms of upregulated DEGs of RCsiu relative to RCsil. e KEGG terms of downregulated DEGs of RCsiu relative to RCsil. f Venn diagram analysis of expressed genes in Rsi, Csi, RCsiu, and RCsil
In contrast, the percentages of coexpressed genes between RCsil (18,884, 86.7% of total expressed genes) and RCsiu (18,884, 92.5% of total expressed genes) were smaller than those of RCseu and RCsel, and RCsil conferred more specific expressed genes than RCsiu (Fig. 3f). In addition, upregulated and downregulated DEGs of RCsiu relative to RCsil were enriched for different KEGG terms, for example, ‘Ribosome’ and ‘Plant pathogen interaction’, respectively (Fig. 3d, e). Intriguingly, we noticed that the pod wall of RCsiu is thicker but softer than that of RCsil. The GO term of upregulated DEGs of RCsil relative to those of RCsiu was significantly enriched for “cell wall”, thus indicating that RCsil is much more lignified than RCsiu (Fig. S6). We next identified 14 upregulated homoeologous gene pairs in RCsil enriched for the “cell wall”, which might explain the more rigid pod wall of RCsil than that of RCsiu (Fig. S6). Furthermore, pod shattering was clearly observed in RCsil, but not RCsiu, during harvest. A previous study showed that the IND gene is required for pod shattering37 and that the RPL gene could prevent replum cells from developing into valve margins38. Analysis of the expression level of the two aforementioned genes revealed that RPL is significantly expressed in Rsi, and the expression level in RCsiu is higher than that of RCsil. However, IND is mainly expressed in Csi and RCsil, which might explain the different phenotypes in pod shattering between RCsiu and RCsil (Fig. S6). Nevertheless, more transcriptome data in the early developmental stages of siliques are required to study these differences. Together, these results elucidate the expression profile differences between the upper and lower sections of siliques of the hybrid.
Genome-wide unbalanced biased expression toward CC in pods and seeds of RrRrCrCr
Homoeolog expression bias refers to the preferential expression of one homoeolog relative to the other in the hybrids22. To study the homoeolog expression bias in RCsiu, RCsim, RCsil, and RCseu, the genes that were expressed in at least one progenitor and where both homoeologs were expressed in Raphanobrassica were analyzed. First, the BBH method was adopted to identify 23,566 orthologous gene pairs between RsRs and C°C° genomes (homoeologous gene pairs of the hybrid subgenomes). Then, a total of 2627, 2939, 2994, and 2594 homoeologous gene pairs were used for expression bias analysis in RCsiu, RCsim, RCsil, and RCseu, respectively. Unbalanced biased expression with a preference toward CC was observed in all four tissues, with a total of 1177 (44.7%), 892 (30.3%), 898 (29.9%), and 1001 (38.6%) homoeologous gene pairs detected in RCsiu, RCsim, RCsil, and RCseu, respectively (Fig. 4). In contrast, the numbers (percentages) of homoeologous gene pairs with RR-biased expression were 799 (30.4%), 627 (21.4%), 642 (21.5%), and 712 (27.5%) in RCsiu, RCsim, RCsil, and RCseu, respectively, which were lower than those of the CC-biased pairs (Fig. 4).

a Homoeolog expression bias analyses of RCsiu, (b) RCsim, (c) RCsil and (d) RCseu. The numbers of homoeolog pairs and their proportions to the total numbers of analyzed pairs are listed. The sizes of the circles in the diploid progenitor RsRs and CoCo indicated the relative expression levels of the orthologs (homoeologs in the hybrid), while relative expression levels of the homoeologs in RrRrCrCr were indicated by the area ratio of the circles. In addition, the numbers in red indicate that the expression patterns of homoeologs in RrRrCrCr maintained parental conditions, while those in blue indicate novel expression bias in the hybrid
Remarkably, more gene pairs showed RR-biased expression in RCsiu and RCseu than in RCsim and RCsil, which might explain why the phenotypes of RCsiu and RCseu share more similarities to R. sativus than those of RCsim and RCsil (Fig. 4, Fig. 1b–d and Fig. S1). In addition, a total of 651 (24.9%), 1420 (48.3%), 1454 (48.6%), and 881 (33.9%) gene pairs showed no bias in RCsiu, RCsim, RCsil, and RCseu of Raphanobrassica, respectively (Fig. 4). Impressively, many homoeologous gene pairs that displayed novel bias were observed in RCsiu (1 172, 44.5%), RCseu (989, 38.2%), RCsim (898, 30.5%), and RCsil (909, 30.3%), among which most of the homoeolog pairs with preexisting biased expression toward RR reverted to biased expression toward CC (Fig. 4). Furthermore, approximately one-third of the respective homoeolog pairs analyzed were maintained under progenitor conditions (Fig. 4). Collectively, the analysis of homoeolog expression bias revealed unbalanced biased expression with a preference toward CC, and more homoeolog gene pairs showed RR-biased expression in RCsiu and RCseu than in RCsim and RCsil. The much more homoeolog gene pairs that show RR-biased expression might explain why the phenotypes of RCsiu and RCseu share more similarities to R. sativus than those of RCsim and RCsil.
Genome-wide expression level dominance biased toward CC in pods and seeds of Raphanobrassica
Expression level dominance (ELD) is another important concept distinct from homoeolog expression bias, which refers to the total expression level of a homoeolog pair in allopolyploids compared to its two progenitors22. To explore the ELD in RCsiu, RCsim, RCsil, and RCseu of Raphanobrassica, we only focused on the genes that are expressed in at least one progenitor and where both homoeologs are expressed in Raphanobrassica, and these gene pairs were classified into 12 categories by comparing the total expression of the homoeolog pairs in RrRrCrCr to those in the progenitors (Fig. 5a).

a ELD in the three sections of pods and RCseu of Raphanobrassica, reflecting the collective expression of homoeolog pairs compared to those in the progenitors. b Relationships between ELD and individual homoeolog expression levels explained the phenomenon of ELD in the four tissues
The results revealed that the numbers of homoeolog pairs displaying CC expression level dominance (ELD-CC, categories II and XI) were much larger than those displaying RR expression level dominance (ELD-RR, categories IV and IX) in RCsiu (621/23.6% vs. 30/1.1%), RCsim (1 169/39.8% vs. 57/1.9%), RCsil (1069/35.7% vs. 79/2.6%), and RCseu (1026/39.6% vs. 45/1.7%) (Fig. 5a). Notably, the homoeolog pairs that display ELD-CC are mainly from category XI, indicating the significant downregulation of RrRr homoeologs compared to those in R. sativus. Additionally, 914 (34.8%), 643 (21.9%), 626 (20.9%) and 813 (31.3%) homoeologous gene pairs showed additivity in RCsiu, RCsim, RCsil and RCseu, respectively (Fig. 5a). Furthermore, a total of 754 (28.7%), 639 (21.7%), 678 (22.6%), and 482 (18.6%) homoeolog pairs displayed transgressive downregulation (categories III, VII, and X) (Fig. 5a). However, only a total of 269 (10.2%), 408 (13.9%), 467 (15.6%), and 210 (8.1%) homoeolog pairs exhibited equivalent expression (“No change” in Fig. 5a) in RCsiu, RCsim, RCsil, and RCseu, respectively. Moreover, the functional categorizations of different sets of homoeolog pairs showing ELD were also studied in GO terms, with three sets of genes (siu-C-ELD, sil-C-ELD, and seu-C-ELD) enriched for “protein heterodimerization activity” and “rRNA binding” (Table S2). However, genes of sim-C-ELD were enriched for photosynthesis (Table S2).
The relationship between ELD and individual homoeolog expression levels in the RCsiu, RCsim, RCsil, and RCseu of Raphanobrassica was also investigated by comparing the individual homoeolog expression levels to those in the diploid progenitors. As expected, the main reason for ELD is that numerous homoeologs or RrRr homoeologs were downregulated compared to those in the diploid progenitors, with 547 (367 + 180, 84.0%), 1 026 (656 + 370, 83.7%), 917 (559 + 358, 79.9%) and 1 026 (670 + 356, 95.8%) homoeolog pairs detected in RCsiu, RCsim, RCsil, and RCseu, respectively (Fig. 5b). Together, these results reveal that the expression level dominance in RCsiu, RCsim, RCsil, and RCseu is toward CC, and the main reason for ELD is due to the downregulated expression of both RrRr and CrCr homoeologs or the downregulated expression of RrRr homoeologs.
Discussion
Hybridization between different species promotes genome evolution and the formation of new crops by genetic recombination, which is usually used to generate novel cultivars with novel genetic traits. Hybrids usually display hybrid vigor (heterosis), disease resistance, and some other traits in future generations12. For example, the Chinese cabbage F1 hybrid Xin No. 3, a popular leafy crop species generated from hybridization between the inbred lines SD and JEY, displays strong heterosis for biomass during vegetative growth and development4. In this study, the intergeneric hybrid derived from the hybridization of R. sativus (RR, 2n = 18) and B. oleracea (CC, 2n = 18) was used for the analysis of mRNA expression profiles. GISH experiments showed that Raphanobrassica has the expected chromosome number through many generations of fertility selection31,32. R. sativus was reported to resist different diseases39. The transfer of the RsRs genome from R. sativus into Raphanobrassica may provide an important resource for disease resistance traits. Our previous study showed the resistance of Raphanobrassica to powdery mildew disease, which may provide an important bridge for disease resistance breeding40. Moreover, plenty of evidence has shown that Raphanobrassica has resistance to clubroot disease, beet cyst nematodes, and effective crossability with Brassica species33. Therefore, Raphanobrassica generated in this work provides a potential new genetic resource for practical use in breeding clubroot-resistant cultivars of Brassica crops. In addition, Raphanobrassica is superior to its two progenitors in dry weight and crude protein content32, which would serve as a useful fodder crop, and the dehiscence characteristic of RCsil makes the siliques easily harvested mechanically. However, how the interaction between the RsRs genome of R. sativus and the C°C° genome of B. oleracea affects chromosome rearrangement in Raphanobrassica remains unclear. Nevertheless, our global gene expression pattern analysis reported here will help the research community broadly understand the consequence of the incorporation of RsRs and C°C° genomes in the silique of Raphanobrassica.
Transcriptome shock in the hybrid refers to extensive changes to patterns of parental gene expression41. In this study, transcriptome shock was observed in all five tissues of RrRrCrCr. A previous study revealed that transcriptome shock might be caused by interspecific hybridization and subsequently ameliorated by genome duplication42. Thus, we speculated that changes in the expression patterns of the diploid F1 hybrid might be more extensive than those of the allotetraploid hybrid when compared to the diploid progenitors. In addition, altered chromatin compaction and histone methylation in the hybrid43, as well as cis- and trans-regulatory divergence between progenitor species44, may also explain the transcriptome shock observed here. Considering that most DEGs in RrRrCrCr are downregulated relative to those of the maternal parent R. sativus, we speculated that chromatin of the RrRr subgenome is much more compact than that of the CrCr subgenome, and this hypothesis could be confirmed by ATAC-seq and Hi-C in future studies.
Homoeolog expression bias has been broadly studied in the past decade13,15,45,46,47. Interestingly, in our study, the silique of Raphanobrassica was split into two parts, and homoeolog expression bias showed a preference for CC. However, more homoeolog gene pairs showed RR-biased expression in RCsiu and RCseu than in RCsim and RCsil. Strikingly, homoeolog expression and induction bias were revealed in Triticum aestivum when bread wheat was infected by the fungal pathogen Fusarium pseudograminearum45. Once infected by the aforementioned fungal pathogen, B and D homoeologs exhibit stronger responses to the infection than A homoeologs, which implies distinctions of homoeolog expression bias under different biotic and abiotic stresses in many resynthesized or natural hybrids. Indeed, some recent studies have also revealed that the expression levels of certain homoeologous genes are altered under heat tolerance, water stress, and iron deficiency in polyploid wheat48,49,50. Hence, homoeolog expression bias of pods and seeds of Raphanobrassica under different stresses still remains to be studied.
ELD has also been widely explored in many resynthesized hybrids13,15,17,51,52. The parental genome toward which ELD of the synthetic hybrid biases is consistent with the similarity of phenotypes between the hybrid and parents. For example, in our study, ELD of pods and seeds of RrRrCrCr showing preference toward CC is consistent with the fact that phenotypes of the whole silique of Raphanobrassica are similar to those of B. oleracea. Hence, we could infer the ELD of the resynthesized hybrid by the similarity of the phenotypes between the progenitors and the hybrid. In addition, there is different ELD biased expression in different organs within the same organism. For example, one previous study revealed that the gene in immature leaves of Raphanobrassica displays genome-wide ELD bias toward RR, which is different from that of the pods and seeds in the present study51. Intrigued, the ELD of different developmental stages of pods and seeds of Raphanobrassica still remains to be explored to comprehensively understand the relationships of global gene expression patterns between the hybrid and its two diploid progenitors.
Collectively, transcriptome shock was observed in the silique of Raphanobrassica due to the incorporation and interaction of the two parental genomes, which might lead to the split of the silique into two parts. The upper and lower sections of siliques of the hybrid exhibit expression profile differences, and the related genes are involved in different biological processes. Many more genes in the silique of Raphanobrassica exhibit homoeolog expression bias and expression level dominance toward B. oleracea than those toward R. sativus. Nonetheless, RCsiu and RCseu have more RR-biased expression genes than RCsim and RCsil, which might explain why the phenotypes of RCsiu and RCseu of the hybrid share more similarities to R. sativus than those of RCsim and RCsil.
In conclusion, our mRNA transcriptome profiles are consistent with the observed phenotypes and unravel the tight correlation between the phenotypes and global gene expression patterns in the hybrid and its parents. Our results reported here provide a good reference to study plant polyploidy and mine potential candidate genes responsible for specific phenotypes. However, whether the phenotypes are tightly correlated with other multiomics data in the synthesized hybrid and its parents remains to be explored.
Materials and methods
Plant materials and sampling
The diploid maternal R. sativus (2n = 2x = 18) and paternal B. oleracea var. alboglabra (2n = 2x = 18) were used as parental lines to generate allotetraploid Raphanobrassica (2n = 4x = 36) by crossing, and the hybrid chromosomes were doubled in the F1 generation by colchicine treatment followed by seed fertility selection32. All of the plant materials were planted in Wuhan, China. Seeds with good fertility were obtained in the F10 generation, and GISH analysis revealed that the hybrid genome contained all 18 chromosomes from RsRs and all 18 chromosomes from C°C°32.
Library construction, Illumina sequencing, read mapping and DEG analysis
Whole pods (Rsi and Csi), seeds (Rse and Cse) of siliques of the diploid progenitors, three sections of the whole pods (RCsiu, RCsim, and RCsil) and all seeds in the two sections of the siliques (RCseu and RCsel) of the hybrid, with three biological replicates of each sample, were obtained at 35 DAF for total mRNA isolation and sequencing library construction.
For RNA-seq, RNA library preparation for each sample was performed according to the manuals provided by Illumina. The aforementioned 27 sequencing libraries were sequenced on the Illumina HiSeq 2000 platform. The raw data were filtered by NGS QC Toolkit53 to obtain high-quality clean reads. The total clean reads of R. sativus and B. oleracea obtained after filtration were mapped to the RsRs and C°C° genomes, respectively, while those of RrRrCrCr were mapped to the integrated genomes of RsRs and C°C° using HISAT software (Table S1)54. We also mapped the clean reads of R. sativus and B. oleracea to the C°C° and RsRs genomes to examine whether erroneous mapping existed in the hybrid, and some of the randomly selected results were further confirmed by RT–qPCR. The novel gene loci were predicted by StringTie55 and annotated by the Pfam database56. The mapping results were also processed by StringTie to obtain FPKM for all 27 samples, and the genes whose FPKM ≥ 1 were identified as expressed. Differentially expressed gene analysis between the 27 samples was conducted using the R package DEseq57, and genes that exhibited a difference of at least twofold with a corrected P value ≤0.05 were regarded as significantly differentially expressed.
GO and KEGG enrichment analysis
GO and KEGG enrichment analyses for the DEGs were conducted using the R package clusterProfiler58 and the KEGG database59, respectively. The GO and KEGG terms of the three ontologies exhibiting padj≤0.05 were regarded as significantly enriched.
Annotation of homoeologous gene pairs in Raphanobrassica
For convenience, we considered that most of the homoeologs remained in a one-to-one relationship after polyploidization within the subgenomes of RrRrCrCr, although the homoeologous relationships are not necessarily one-to-one60. Based on this, we adopted the BBH method to infer homoeologs61, although this method has inherent drawbacks62. Thus, as shown in Fig. S7, the 43,882 gene models of C°C° and the 48,203 gene models of RsRs were blasted against each other using the blastn program63 with a search cutoff e-value of 1e−10 to identify the orthologous gene pairs, which were termed homoeologous gene pairs in the hybrid. Gene pairs with a sequence identity > =90% were regarded as orthologous gene pairs in the two progenitors or homoeologous gene pairs in the hybrid.
Analysis of homoeolog expression bias and expression level dominance
In the process of homoeolog expression bias and ELD analyses, we only paid attention to the genes that were expressed in at least one progenitor and where both homoeologs were expressed in Raphanobrassica. Student’s t-test (P < 0.05) was used to compare the expression level of each homoeologous gene pair in the two progenitors (RsRs vs. C°C°) and the Raphanobrassica hybrid (RrRr vs. CrCr) in the process of homoeolog expression bias according to previously described methods13. During the analyses of ELD in different tissues, the collective expression level of a homoeologous gene pair in the Raphanobrassica hybrid was compared to that of the diploid progenitors, that is, (RrRr + CrCr) vs. RsRs and (RrRr + CrCr) vs. C°C°, using Student’s t-test (P < 0.05) as described previously13. Furthermore, twelve possible bins were classified according to Yoo et al.52.
Collinearity analysis
The protein, CDS, and mRNA FASTA files and GFF files for B. oleracea and R. sativus were retrieved from the NCBI genome database, and only genes of R. sativus from the nine longest scaffolds were used for analysis. Moreover, only the first transcript was used when the gene had more than one transcript. Homologous genes within the self-genome or cross-genome were obtained using BLASTP (blast−2.9.0+)64 with a cutoff e-value of 1e−10, and only the top five hits were used for downstream collinearity analysis using the MCSanX package65.
RT–qPCR validation
The expression level of mRNA was detected in the nine tissues of R. sativus, B. oleracea, and hybrids using quantitative reverse transcription-PCR (RT–qPCR). For mRNA expression level detection, 1 μg of total RNA was reverse-transcribed using SuperScript III Reverse Transcriptase (Invitrogen) and oligo (dT)18 according to the manufacturer’s protocol. The qPCR experiment was carried out using an ABI 7300 (ABI), and each reaction was performed in triplicate. U6 RNA was set as an internal reference gene for mRNA expression detection. The primers for mRNA RT–qPCR are listed in Table S3.
Data availability
Sequencing data of R. sativus, B. oleracea, and the hybrid Raphanobrassica were deposited in the SRA Database in NCBI (Accession numbers: SRR12191747, SRR12191748, SRR12191749, SRR12191750, SRR12191751, SRR12191752, SRR12191753, SRR12191754, SRR12191756, SRR12191757, SRR12191758, SRR12191759, SRR12191760, SRR12191761, SRR12191762, SRR12191763, SRR12191764, SRR12191765, SRR12191766, SRR12191767, SRR12191768, SRR12191769, SRR12191770, SRR12191771, SRR12191772, SRR12191773).
References
Brenchley, R. et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491, 705–710 (2012).
Wang, M. et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 51, 224–229 (2019).
Chalhoub, B. et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953 (2014).
Li, P. et al. Genome-wide analysis of changes in miRNA and target gene expression reveals key roles in heterosis for Chinese cabbage biomass. Horticulture Res. 8, 39 (2021).
Jackson, S. & Chen, Z. J. Genomic and expression plasticity of polyploidy. Curr. Opin. Plant Biol. 13, 153–159 (2010).
Sosnowska, K. et al. Chromosome instabilities in resynthesized Brassica napus revealed by FISH. J. Appl. Genet. 61, 323–335 (2020).
Schiavinato, M., Bodrug‐Schepers, A., Dohm, J. C. & Himmelbauer, H. Subgenome evolution in allotetraploid plants. Plant J. 106, 672–688 (2021).
Qiu, T., Liu, Z. & Liu, B. The effects of hybridization and genome doubling in plant evolution via allopolyploidy. Mol. Biol. Rep. 47, 5549–5558 (2020).
Glombik, M., Bačovský, V., Hobza, R. & Kopecký, D. Competition of parental genomes in plant hybrids. Front. Plant Sci. 11, 200 (2020).
Voshall, A. & Moriyama, E. N. Next-generation transcriptome assembly and analysis: Impact of ploidy. Methods 176, 14–24 (2020).
Ni, Z. et al. Altered circadian rhythms regulate growth vigour in hybrids and allopolyploids. Nature 457, 327–331 (2009).
Leitch, A. & Leitch, I. Genomic plasticity and the diversity of polyploid plants. Science 320, 481–483 (2008).
Wu, J. et al. Homoeolog expression bias and expression level dominance in resynthesized allopolyploid Brassica napus. BMC Genomics 19, 586 (2018).
Zhang, L. et al. Integration of metabolome and transcriptome reveals flavonoid accumulation in the intergeneric hybrid between Brassica rapa and Raphanus sativus. Sci. Rep. 9, 18368 (2019).
Wu, Y. et al. Transcriptome shock in an interspecific F1 triploid hybrid of Oryza revealed by RNA sequencing. J. Integr. Plant Biol. 58, 150–164 (2016).
Landis, J. B., Kurti, A., Lawhorn, A. J., Litt, A. & McCarthy, E. W. Differential gene expression with an emphasis on floral organ size differences in natural and synthetic polyploids of Nicotiana tabacum (Solanaceae). Genes 11, 1097 (2020).
Li, A. et al. mRNA and small RNA transcriptomes reveal insights into dynamic homoeolog regulation of allopolyploid heterosis in nascent hexaploid wheat. Plant Cell 26, 1878–1900 (2014).
Shahzad, K. et al. Global insights into homoeolog gene expression in upland cotton under intraspecific hybridization. https://doi.org/10.21203/rs.3.rs-234267/v1 (2021).
Xiong, Z., Gaeta, R. T. & Pires, J. C. Homoeologous shuffling and chromosome compensation maintain genome balance in resynthesized allopolyploid Brassica napus. Proc. Natl Acad. Sci. 108, 7908–7913 (2011).
Szadkowski, E. et al. Polyploid formation pathways have an impact on genetic rearrangements in resynthesized Brassica napus. N. Phytologist 191, 884–894 (2011).
Zhang, L. et al. Characterization and expression profiles of miRNAs in the triploid hybrids of Brassica napus and Brassica rapa. BMC Genomics 20, 649 (2019).
Grover, C. et al. Homoeolog expression bias and expression level dominance in allopolyploids. N. Phytologist 196, 966–971 (2012).
Li, M., Wang, R., Wu, X. & Wang, J. Homoeolog expression bias and expression level dominance (ELD) in four tissues of natural allotetraploid Brassica napus. BMC Genomics 21, 330 (2020).
Sigel, E. M., Der, J. P., Windham, M. D. & Pryer, K. M. Expression level dominance and homeolog expression bias in recurrent origins of the allopolyploid fern Polypodium hesperium. Am. Fern J. 109, 224–247 (2019).
Gallagher, J. P., Grover, C. E., Hu, G., Jareczek, J. J. & Wendel, J. F. Conservation and divergence in duplicated fiber coexpression networks accompanying domestication of the polyploid Gossypium hirsutum L. G3: Genes, Genomes, Genet. 10, 2879–2892 (2020).
Hu, G. et al. Homoeologous gene expression and co-expression network analyses and evolutionary inference in allopolyploids. Brief. Bioinforma. 22, 1819–1835 (2020).
Zhao, N. et al. Evolution of homeologous gene expression in polyploid wheat. Genes 11, 1401 (2020).
Chen, Z. J. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu. Rev. Plant Biol. 58, 377–406 (2007).
Nugroho, A. B. D., Han, N., Pervitasari, A. N., Kim, D.-H. & Kim, J. Differential expression of major genes involved in the biosynthesis of aliphatic glucosinolates in intergeneric Baemoochae (Brassicaceae) and its parents during development. Plant Mol. Biol. 102, 171–184 (2020).
Lee, S.-S. et al. Properties of self-sterile but cross-fertile allopolyploids synthesized between Brassica rapa and Raphanus sativus. Horticul. Environ., Biotechnol. 61, 163–171 (2020).
Chen, H. & Wu, J. Characterization of fertile amphidiploid between Raphanus sativus and Brassica alboglabra and the crossability with Brassica species. Genet. Resour. Crop Evol. 55, 143–150 (2008).
Chen, H., Wu, J., Cheng, Y. & Liu, C. Development and GISH analysis of amphidiploid Raphanobrassica between Raphanus sativus and Brassica alboglabra. Acta Agronomica Sin. 32, 1117 (2006).
Zhan, Z. et al. Cytological and morphological analysis of hybrids between Brassicoraphanus, and Brassica napus for introgression of clubroot resistant trait into Brassica napus L. PLoS ONE 12, e0177470 (2017).
Zhang, Y. et al. Suppressing type 2C protein phosphatases alters fruit ripening and the stress response in tomato. Plant Cell Physiol. 59, 142–154 (2018).
Howell, S. H. Endoplasmic reticulum stress responses in plants. Annu. Rev. Plant Biol. 64, 477–499 (2013).
Kotak, S., Vierling, E., Bäumlein, H. & von Koskull-Döring, P. A novel transcriptional cascade regulating expression of heat stress proteins during seed development of Arabidopsis. Plant Cell 19, 182–195 (2007).
Zhai, Y. et al. CRISPR/Cas9-mediated genome editing reveals differences in the contribution of INDEHISCENT homologues to pod shatter resistance in Brassica napus L. Theor. Appl. Genet. 132, 2111–2123 (2019).
Roeder, A. H., Ferrándiz, C. & Yanofsky, M. F. The role of the REPLUMLESS homeodomain protein in patterning the Arabidopsis fruit. Curr. Biol. 13, 1630–1635 (2003).
Wang, X. et al. Resistance gene analogue isolation and RGA-based marker development for identifying downy mildew resistance in radish (Raphanus sativus L.). Euphytica 198, 289–303 (2014).
Alkooranee, J., Liu, S., Aledan, T., Yin, Y. & Li, M. First report of powdery mildew caused by Erysiphe cruciferarum on Brassica napus in China. Plant Dis. 99, 1651–1651 (2015).
Hegarty, M. J. et al. Transcriptome shock after interspecific hybridization in Senecio is ameliorated by genome duplication. Curr. Biol. 16, 1652–1659 (2006).
Wang, J. et al. Genomewide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics 172, 507–517 (2006).
Zhu, W. et al. Altered chromatin compaction and histone methylation drive non-additive gene expression in an interspecific Arabidopsis hybrid. Genome Biol. 18, 157 (2017).
Shi, X. et al. Cis-and trans-regulatory divergence between progenitor species determines gene-expression novelty in Arabidopsis allopolyploids. Nat. Commun. 3, 950 (2012).
Powell, J. J. et al. The defence‐associated transcriptome of hexaploid wheat displays homoeolog expression and induction bias. Plant Biotechnol. J. 15, 533–543 (2017).
Zou, X. et al. Genome evolution in Oryza allopolyploids of various ages: Insights into the process of diploidization. Plant J. 105, 721–735 (2020).
Zhang, Z. et al. Homoeologous exchanges occur through intragenic recombination generating novel transcripts and proteins in wheat and other polyploids. Proc. Natl Acad. Sci. 117, 14561–14571 (2020).
Eva, E., Ragupathi, N. & Grant, N. P. Genome-wide analysis of the HSP101/CLPB gene family for heat tolerance in hexaploid wheat. Sci. Rep. 10, 3948 (2020).
Derakhshani, B. et al. Comparative analysis of root transcriptome reveals candidate genes and expression divergence of homoeologous genes in response to water stress in wheat. Plants 9, 596 (2020).
Wang, M., Gong, J. & Bhullar, N. K. Iron deficiency triggered transcriptome changes in bread wheat. Comput Struct. Biotechnol. J. 18, 2709–2722 (2020).
Ye, B., Wang, R. & Wang, J. Correlation analysis of the mRNA and miRNA expression profiles in the nascent synthetic allotetraploid Raphanobrassica. Sci. Rep. 6, 37416 (2016).
Yoo, M., Szadkowski, E. & Wendel, J. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 110, 171–180 (2013).
Patel, R. K. & Jain, M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE 7, e30619 (2012).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Nat. Precedings https://doi.org/10.1038/npre.2010.4282.1 (2010).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics: a J. Integr. Biol. 16, 284–287 (2012).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Glover, N. M., Redestig, H. & Dessimoz, C. Homoeologs: what are they and how do we infer them? Trends Plant Sci. 21, 609–621 (2016).
Kuo, T. C., Hatakeyama, M., Tameshige, T., Shimizu, K. K. & Sese, J. Homeolog expression quantification methods for allopolyploids. Brief. Bioinforma. 21, 395–407 (2020).
Dalquen, D. A. & Dessimoz, C. Bidirectional best hits miss many orthologs in duplication-rich clades such as plants and animals. Genome Biol. Evolution 5, 1800–1806 (2013).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49–e49 (2012).
Acknowledgements
This research was supported by the National Key Research and Development Program of China (2017YFD0101701) and the National Natural Science Foundation of China (Grant No. 31540083).
Author information
Authors and Affiliations
Contributions
M.L. conceived the idea, designed the study, and revised the manuscript. L.Z., H.H., and J.W. performed the experiments. J.H. performed the bioinformatic analysis. L.Z. and J.H. wrote the manuscript and contributed equally to this work. All the authors have reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, L., He, J., He, H. et al. Genome-wide unbalanced expression bias and expression level dominance toward Brassica oleracea in artificially synthesized intergeneric hybrids of Raphanobrassica. Hortic Res 8, 246 (2021). https://doi.org/10.1038/s41438-021-00672-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-021-00672-2
This article is cited by
-
Transgressive and parental dominant gene expression and cytosine methylation during seed development in Brassica napus hybrids
Theoretical and Applied Genetics (2023)
