
Abstract
The Rosaceae family has striking phenotypic diversity and high syntenic conservation. Gillenia trifoliata is sister species to the Maleae tribe of apple and ~1000 other species. Gillenia has many putative ancestral features, such as herb/sub-shrub habit, dry fruit-bearing and nine base chromosomes. This coalescence of ancestral characters in a phylogenetically important species, positions Gillenia as a ‘rosetta stone’ for translational science within Rosaceae. We present genomic and phenological resources to facilitate the use of Gillenia for this purpose. The Gillenia genome is the first fully annotated chromosome-level assembly with an ancestral genome complement (x = 9), and with it we developed an improved model of the Rosaceae ancestral genome. MADS and NAC gene family analyses revealed genome dynamics correlated with growth and reproduction and we demonstrate how Gillenia can be a negative control for studying fleshy fruit development in Rosaceae.
Similar content being viewed by others

The Rosaceae family displays wide phenotypic diversity especially with regard to fleshy fruit forms1. All fleshy fruiting crops putatively arose from dry-fruiting ancestors2 and although most of the 3000 species within Rosaceae are dry-fruiting, they can be distilled down to two types, achenes and follicles, thus providing a common basis for comparative analyses. Comparisons between dry and fleshy fruiting relatives provide an alternative approach to study fruit development. Comparative genomics of almond and peach highlighted the potential of the transposable element landscape to alter the fate of fruit tissues3 and a comparison of woodland (Fragaria vesca) and pink barren (Potentilla micrantha) strawberries underscored the importance of MADS-box regulation of embryo and seed development for flesh development4. Despite the economic importance of pome fruits (Malus domestica (apple) and Pyrus spp. (pear)), no closely related dry-fruiting model is currently available.
Apple and pear belong to the Maleae tribe. The origin of this tribe proposes that ~50 Mya an ancestral species with x = 9 underwent autopolyploidisation, aneuploidy, and diploidisation to give rise to apple and pear (2n = 2x = 34) and sister taxa2,5,6,7. It is proposed that the ancestral species was similar to extant genus Gillenia. The genome of Gillenia remained unduplicated (2n = 2x = 18) and therefore provides a simpler foundation to untangle complexities of Maleae whole-genome duplication (WGD) as well as to improve prediction of genome configurations of the Rosaceae common ancestor.
The phenotype and genome of G. trifoliata has potential to provide a ‘rosetta stone’ for translational science within Rosaceae. The genome complement of G. trifoliata (x = 9) is similar to most Rosaceae species (x = 7, 8, 9) and represents the chromosome complement of the putative ancestor to all Rosaceae8,9,10,11. Currently no chromosome-level, annotated assemblies are available within Rosaceae with ancestral genome complement (x = 9), as Dryas drummondii and Purshia tridentata are both unannotated drafts12. All high-quality Rosaceae genomes sequenced to date are for either fleshy-fruited species or dry achene-fruiting species with fleshy accessories. A sequenced genome for a dry follicle-fruiting species, the ancestral fruit type of the second-largest subfamily Amygdaloideae (e.g. apple, peach, cherry), is currently unavailable. The growth habit of the largest Rosaceae subfamily, Rosoideae, is predominantly herbaceous, with some form of asexual propagation such as rhizomes, stolons, runners or suckers2. The rhizomatous annual-fruiting perennial herb or sub-shrub growth habit of G. trifoliata will enable comparative studies of shoot architecture, floral induction and biennial fruit bearing.
Here, we provide genomic and phenotypic resources to facilitate the use of G. trifoliata as a model species for comparative analyses. The phenotype of G trifoliata is described in terms of phenological growth stages using a Biologische, Bundesanstalt, Bundessortenamt, and Chemical Industry (BBCH) scale to assist phenotypic comparisons. A high-quality whole-genome sequence for G. trifoliata has been constructed with genome assembly and annotation metrics consistent with its phylogenetic position within the Rosaceae. Analysis of MADS and NAC transcription factor families reveal genome dynamics that correlate with differences in growth and reproductive phenotype between G. trifoliata and Rosaceae species. Lastly, a study of selection pressure upon SEPALLATA1/2-like MADS8 orthologues with known intra-familial roles in fruit flesh development, demonstrates the potential for research using G. trifoliata to reveal new insights into fruit flesh development.
Results
Phenology of Gillenia trifoliata within a BBCH framework
To enable intra-familial comparisons, phenological stages were described within a BBCH framework. The BBCH scale provides a universal code to facilitate standardisation of key plant development phases13. Scales are established for model plants and key horticultural crops14,15.
G. trifoliata is a perennial herb which grows, reproduces, and dies-back annually to a woody rhizome that then undergoes winter dormancy. The growth cycle of G. trifoliata can be described in nine phenological growth stages over time (Fig. 1a). Each rhizome bears multiple perennating buds that give rise to shoot systems comprised of both main and lateral growth that are mostly reproductive (Fig. 1b). Rhizome shoots differ in vigour, as primary shoots (~25% of shoots) have greater vigour than secondary shoots in terms of length, node number, axillary growth, and reproductive or vegetative fate (Supplementary Fig. 1). The difference in vigour was borne out by steeper growth in the first phase of a double-sigmoidal growth curve (Fig. 1b). To manage the growth complexity and maximise utility of a phenological tool, we describe two levels of BBCH scale: a shoot-level scale to describe whole-plant development, and an organ-level scale for reproductive development. The shoot-level scale accommodates growth complexity by being extendable for differences in vigour. Detailed description of the growth stages is provided in Supplementary Note S.1, Supplementary Figs. 1‒9, and Supplementary Tables 1 and 2.

a Duration and timing of primary growth stages over time. b (top left) Schematic arrangement of primary (red) and secondary (pink) reproductive and vegetative (green) shoots of a G. trifoliata plant during bud development (top right, PGS 0), and growth curves of primary and secondary reproductive shoots showing difference in vigour with key shoot-level growth stages indicated. c Key stages during whole plant development covered by the shoot-level BBCH scale with correlating growth stages given for each stage. d Key stages of the reproductive organ-level BBCH scale, from left, inflorescence emergence (stage 55), end of inflorescence emergence (stage 59), full flowering (stage 65), early fruit development (stage 71), early fruit senescence (stage 85), full fruit senescence (stage 89)
Whole-plant development begins with main shoot growth from rhizome buds occurring over the first 30 days after dormancy (DAD, BBCH stages 0/1/3, Fig. 1c). Lateral shoot development (BBCH stage 2) and flowering (BBCH stages 5/6) occur concurrently, with flowering concentrated at the distal ends of both main and lateral shoots (Fig. 1c, d). A long period of fruit development (~110 days, BBCH stage 7) is followed by late-summer fruit senescence (BBCH stage 8). Vegetative senescence (BBCH stage 9) begins near the end of flowering and continues throughout the season.
The reproductive organ-level scale is independent of shoot position, to aid comparative sample collection. Inflorescence emergence begins (stage 51) after shoots have emerged, first individual flowers are visible (stage 55) by 30 DAD, and first flowers reach the equivalent apple “balloon” stage (stage 59) by 42 DAD. Flowers open (stage 60) with unfurling of the petal tube and proceed rapidly in 2‒3 days to fully open (stage 65), after which stamens darken, petals abscise and hypanthium remains attached. After pollination (stage 70), visible follicle growth is evident by 6 days after pollination (DAP, stage 71). Final fruit size is reached quickly at ~21 DAP (stage 72) and seed coat colour changes to burnt orange (stage 79). Fruit growth to maturity and beginning of fruit senescence (stage 80) takes ~130 DAP, similar to apple, and fruit senescence ends (stage 89) at ~160 DAP.
Genome assembly and annotation
The genome size of G. trifoliata was estimated at 321.6 Mb by flow cytometry (Supplementary Fig. 10, Supplementary Table 3). The nuclear genome was firstly sequenced and assembled from 10X Chromium linked Illumina short reads, which produced two haplotype genome assemblies. One haplotype displayed improved assembly statistics, with a total size of 269 Mb, N50 of 847.9 kb and excellent BUSCO16 completeness (96.7% with a duplication rate as low as 2.6%). Oxford Nanopore Technology (ONT) long-read and long-range Illumina Hi-C short-read technologies were later used to improve the continuity and completeness of this haplotype assembly through post-scaffolding and anchoring scaffolds to chromosomal level, respectively (Table 1; Supplementary Fig. 11). The final assembly size was 296.3 Mb, similar to the genome size of peach17 (Table 2), and containing only 0.25% of gaps (N%). A total of 272.1 Mb were anchored to nine pseudo-chromosomes with lengths ranging from 22.3 to 46.3 Mb (Supplementary Table 4). Aligning 10X Illumina short reads to the assembly showed an overall alignment rate of 86.3%. BUSCO analyses16 predicted a high-quality assembly with completeness of 98.0% (95.6% unique and 2.4% duplicated) with 0.6% fragmented and 1.8% missing genes. K-mer analysis using 10X Illumina short reads estimated genome heterozygosity at 0.565% (Supplementary Fig. 12).
Genome synteny supports ancestral predictions
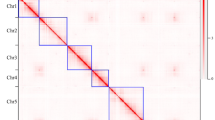
Comparative genome mapping was performed between G. trifoliata and apple18. Dot plot analysis using syntenic blocks with a minimum length of 5 kb sharing at least 70% nucleotide identity demonstrated a high level of conserved synteny between G. trifoliata and apple and pear (Fig. 2a; Supplementary Fig. 13a, b). Syntenic chromosomal regions in G. trifoliata typically hit two homologous regions in apple and pear19 as expected because of the Maleae WGD event. Seven G. trifoliata chromosomes were completely collinear with apple chromosomes from end-to-end: G. trifoliata Chr01, Chr02, Chr03, Chr05, Chr06, Chr07 and Chr08 with apple and pear Chr05/10, Chr03/11, Chr09/17, Chr12, Chr06, Chr07 and Chr08, respectively. Apple Chr13/16 have near-complete collinearity with G. trifoliata Chr04, except for their distal ends. Syntenic alignments of partial chromosomes replicate and validate the hypothesised reconstruction of chromosomes of the Maleae ancestor prior to WGD as described by Velasco et al.5. The only addition is conserved synteny between the proximal end of apple Chr04 and the distal end of G. trifoliata Chr04, which was not previously resolved.

a Circos plot of synteny between the genomes of G. trifoliata (Gtr) and apple (Mdo)18. Each connecting line links two homological regions longer than 10 kb. b Model for the evolution of present day Rosaceae species from hypothetical ancestor. The ancestral chromosomes are represented in nine colours, while chromosomes of descendants, peach17 and raspberry20, are coloured according to ancestral origin. Estimates for the number of fissions and fusions are given at each node. Dashed region of raspberry Chr01 represents area for which ancestral origin could not be determined
Synteny was assessed against the peach17 and Rubus occidentalis (black raspberry)20 genomes using orthologous blocks with a minimum length of 2 and 1 kb, respectively, sharing at least 70% nucleotide identity. A high degree of synteny was detected against both genomes and, unlike the syntenic map of G. trifoliata versus apple, all G. trifoliata chromosomes hit only one orthologous region in both peach and raspberry (Supplementary Fig. 13c, d). Despite extensive rearrangements, syntenic conservation between the distal end of G. trifoliata Chr04 with peach Chr01 and raspberry Chr04 supports the assembly of G. trifoliata Chr04. Two inversions were detected within the distal halves of G. trifoliata Chr02 and Chr03, which were common to all four comparative species, suggesting G. trifoliata -only events. A single small region within the proximal half of G. trifoliata Chr01 had consistently low nucleotide identity across all four species, indicating putative divergence unique to G. trifoliata.
Ancestral genome reconstructions have been proposed for the Rosaceae family. The G. trifoliata genome was compared with the reconstructions of Jung et al.10 and showed fusion and fission events predicted for Malus Chr12/04/14 (G. trifoliata Chr05) which would have instead occurred respectively prior to, and after Maleae WGD. G. trifoliata therefore provides a simplified representation of the Malodae super-tribe. Combined with the conservation of synteny between Gillenia, peach and raspberry, a new model for the Rosaceae ancestral genome was developed. Forty-four syntenic blocks identified across all species were used to reconstruct a model of the nine chromosomes of ancestral Rosaceae (Fig. 2b, Supplementary Fig. 14). Both the new model and the model of Jung et al.10 required the same number of rearrangements to form raspberry (Rosoideae). However, the new model improved upon the model of Jung et al.10 in requiring fewer fissions and fusions to form Prunus (Amygdaloideae; Supplementary Fig. 15).
Comparison of the transposable element (TE) landscape
In total, 47.2% of the G. trifoliata genome assembly was annotated as repeat elements, mainly from long terminal repeat (LTR) and terminal inverted repeat (TIR) classes (Table 2, Supplementary Table 5). Across the nine chromosomes, Chr01 and Chr08 had the highest and lowest TE proportions respectively (Supplementary Tables 6–7). Chr01 and Chr08 were found to have respectively low and high syntenic conservation in dot plot analyses with Amygdaloideae species (Supplementary Fig. 13). The density of annotated TEs on Chr01 was highest at the proximal end and correlated with a high density of annotated Gypsy class TEs (Fig. 3a). Compared with other Rosaceae, G. trifoliata had proportionally higher counts of PIF_Harbinger and hAT classes, and proportionally lower counts of Gypsy and LTR_unknown classes, in both annotated and intact comparisons (Supplementary Fig. 16). Combined with the observation of a high density of annotated Gypsy classes in proximal Chr01 (Fig. 3a), degeneration of this TE type may correlate with the low syntenic conservation with apple and pear in this region.

a Circos plot of features in G. trifoliata genome. Tracks from outer to inner circles indicate: chromosomes coloured by ancestral origin, gene density (each y-axis partition represents 100 k), the density of all annotated transposable elements (TE; each y-axis partition represents 200 k), the density of annotated Gypsy and Copia elements (each y-axis partition represents 50 k). The window size for all density tracks was 1 Mb. Orthologous cluster analysis (b, c) with a summary of orthologous clusters and singletons (b) and Venn-diagram of relationships between orthologous clusters (c) found in a comparison of annotated proteins from six Rosaceae species: G. trifoliata, apple22, pear32, peach17, strawberry72, and raspberry20
The apple HODOR sequence (KX869746.1) of 9,716 nts was used to verify TEs detected by the Extensive de novo TE Annotator (EDTA) pipeline. An abundance of BLAST high-scoring segment pairs (HSPs) between HODOR and G. trifoliata TEs was found, but all TEs belonged to the LTR type. However, the aligned length of HSP was around 1 kb or less, with sequence identity ranging between 80.0% and 91.6%, which indicated that the HODOR repeat putatively appeared after speciation between G. trifoliata and the apple and pear ancestor.
Comparison of the gene landscape
The genic landscape of the G. trifoliata genome was compared to those of other Rosaceae (Table 2). A total of 28,847 protein-coding genes were predicted in G. trifoliata, similar to peach, with the difference in gene density reflected in the higher TE percentage in G. trifoliata. The number of high confidence genes in apple and pear, compared with G. trifoliata, was consistent with the rapid loss of most duplicated genes in ancient polyploid plant genomes21. The mean gene length in G. trifoliata of 2594 bps was shorter than those of the Rosaceae species under comparison. A total of 514 transfer ribonucleic acids (tRNAs) were detected in G. trifoliata, which were most abundant on Chr08 at 2.13 tRNAs/Mb, and in notably high abundance on contig_1706_RaGOO (Supplementary Table 8).
Orthologous cluster analysis was performed with predicted proteins from G. trifoliata, apple (GDDH13), pear (Pbe-SD), peach, strawberry, and raspberry (Fig. 3b, c). Orthologous cluster analysis was previously shown to be a useful metric of annotation quality19. The number of proteins, clusters, and singletons and distribution of clusters for the G. trifoliata protein set was comparable to those of Rosaceae species of interest, relative to its phylogenetic position. Gene Ontology (GO) enrichment was assessed for clusters of G. trifoliata only and all-but-G. trifoliata clusters, and no GO terms were found to be enriched (Supplementary Table 9).
Genome dynamics related to growth habit
Changes to gene family size provide important sources of genetic variation for evolution21. Gene family sizes between G. trifoliata and apple were assessed to identify genome dynamics correlating with differences in phenotype. The MADS transcription factor family is known to control many aspects of growth and development and was mined from G. trifoliata and two recent apple genomes18,22. The G. trifoliata MADS family was similar in size to that of peach (Table 3). Between subfamilies, Type II MADS had a bias toward retention of both homoeologous gene pairs, whereas the Type I MADS subfamily was similar in size between apple, G. trifoliata and peach (Table 3; Supplementary Fig. 17), demonstrating contrasting evolutionary dynamics observed previously23.
Two notable clade contractions in G. trifoliata were observed for Type II MADS, which may correlate with differences in growth habit (Fig. 4a). In the ANR1 clade, two homoeologous loci were observed in apple, whereas the orthologous locus was absent in G. trifoliata. In Arabidopsis, ANR1 controls lateral and primary root development24. Comparison of the loci at the genomic level identified a large inversion in G. trifoliata bordering the site where the ANR1 locus should be, suggesting disruption of this locus during genomic rearrangements (Supplementary Fig. 18a). Investigation of the ANR1 clade across the Rosaceae found an absence of ANR1-like sequences in strawberry and raspberry annotations, whereas multiple annotations were observed in pear and single annotations were present in peach and rose (Supplementary Fig. 18b), suggesting a correlation between ANR1 abundance and growth habit, given Rosa contains both shrub and tree habits. In apple, MdSOC1-like was linked to a QTL on Chr01 for biennial bearing25 and resides on one of two homoeologous loci (Chr01/07) that each contain three SOC1-like genes in tandem. In annual-bearing G. trifoliata, only one complete and two pseudogenes were detected at the orthologous locus.

Phylogenetic trees of (a) Type II MADS and (b) NAC transcription factors from apple (Md), Gillenia, and Arabidopsis. Ratios of two homoeologous apple and one orthologous Gillenia genes highlighted in red font. Clade expansions of interest shaded green, putative Rosaceae-specific NAC clade shaded yellow. Maximum likelihood bootstrap values from 100 datasets are shown at branches. Prefixes: Gtr, Gillenia trifoliata; MDXXG, apple GDDH1322; MDXX_HF, apple HFTH118; AT, Arabidopsis thaliana; Soly, tomato. MADS subfamily names follow standard convention and27 for Maleae-specific MIKCC-I and –II, letters in parentheses denote floral homeotic classes. NAC subfamily names follow81,82. Apple fruit development NACs in ANAC1 tandem array (arrow) and fruit ripening clades (arrowhead) identified in ref. 35
A notable asymmetric clade contraction in apple was observed in the StMADS11/SVP clade, where five genes exist on G. trifoliata and apple Chr08, but only two on apple Chr15, demonstrating a departure from retention of duplicated genes post-WGD strongly observed in this gene subfamily. In apple, this clade is associated with the regulation of dormancy26. Like apple, G. trifoliata has a strong dormancy requirement for optimum growth (Supplementary Note 1.2), which correlates with the near-equivalent number of dormancy-associated MADS in apple. In addition, two clades putatively apple- and pear-specific, MIKCC-I and -II, showed different evolutionarily dynamics with MIKCC-II missing entirely from G. trifoliata. In apple, the expression of MIKCC-II genes was similar to that of SVP-genes with known involvement in dormancy regulation27.
Genome dynamics related to fruit development
G. trifoliata bears a dry follicetum (aggregate of follicles), which develops from an ovary separated from the hypanthium. Key differences from apple include ovary and hypanthium fusion, development of fleshy tissues, and ripening. Gene family analysis was centred on TALE and NAC, and MADS transcription factors, which have roles in organ boundary and floral development, respectively, and expansins with roles in ripening.
Following WGD, transcription factors are frequently retained, to maintain protein stoichiometry or gene dosage balance21. A 2:1 apple:Gillenia ratio dominated the TALE family (Supplementary Fig. 19), and conspicuous amongst this was a 1:1 ratio within a clade related to BELLRINGER (BLR). In Arabidopsis, BLR controls organ boundary, floral and fruit patterning, inflorescence stem development, and is a ubiquitous hub protein in floral gene regulatory networks28. In apple, BLR-like MD07G1205600 (Supplementary Table 10 for MDP to GDDH13/HFTH1 conversions) is highly expressed in floral buds treated with cytokinin to induce floral transition29 and is also expressed in stem, floral and fruit tissues30. The 1:1 ratio in the BLR-like clade was found to be conserved in domesticated and crab apple18,22,31 and pear19,32 genomes, suggesting importance for pome-fruiting species. A 1:1 ratio was also observed in the NAC CUC3 clade (Fig. 4b), which sits within a subfamily dominated by 2:1 ratios; however, this was conserved only in domesticated apple genomes18,22. CUC3 controls organ boundary development and functions upstream of BLR33.
In G. trifoliata, 110 NACs were identified, similar to 112 in strawberry34 and 115 in peach (Table 3). In apple, 196 unique NACs were detected using two recent genome annotations18,22 (Supplementary Table 11 for MDP to GDDH13/HFTH1 conversions). With few exceptions, a 2:1 apple:Gillenia ratio dominated, including in clades with known roles in organ boundary development and ripening (e.g. Ia/CUC3, Va/NAP, Vb/SNAC; Fig. 4b). Notable exceptions included a clade putatively Rosaceae-specific (Fig. 4b ‘ROS’), which may be subject to evolutionary pressures to maintain a constant overall family size, similar to Type I MADS, as 31 NACs were identified in Gillenia and strawberry, and 33 in apple despite WGD (Supplementary Fig. 20a). Also notable was a clade expansion related to a tandem gene array in subfamily IVa/ANAC1. In the array Gillenia has six NACs on Chr07, while apple has 20 and nine NACs respectively on syntenic Chr01 and Chr07. In strawberry this locus has 10 NACs (Supplementary Fig. 20b) suggesting loss of NACs in G. trifoliata and duplication of the entire 10-NAC array on apple Chr01. MD07G1162700 is situated within the Chr07 tandem array (Fig. 4b, arrow) and was recently associated with ethylene/auxin cross-talk for apple fruit development and ripening35. In strawberry, all 10 NACS are expressed in fruit, but consistent with its non-climacteric physiology, were mostly down-regulated in red versus green fruit34. Fewer tandemly arrayed NACs in G. trifoliata may correlate with its dry-fruit physiology. Tandem gene arrays were found to be frequently collapsed in a comparison of the Rubus v1 and v3 genome assemblies20, therefore the assembly of G. trifoliata loci for SOC, TERN, and ANAC1 clades was checked against raw ONT reads. SOC and TERN loci showed >95% identity between assembly and raw ONT reads, whereas the ANAC1 locus showed some evidence for possible collapse. However, error-correction and reannotation of the ANAC1-associated ONT contig, revealed only one additional NAC as a result of the alternate genome arrangements (Supplementary Fig. 21).
In the expansin gene family, clades found to be associated with ripening in apple and pear36 (Supplementary Fig. 22) were found to have a dominant 2:1 apple:Gillenia ratio, suggesting that, despite their role in ripening, selective pressures have not resulted in isolated clade expansions outside WGD. Expansins were of note as a difficult family for gene annotation programs to detect37; however, nearly all computer-generated annotations for G. trifoliata were full models.
Lastly, floral homeotic Type II MADS, SQUA (A), AG (C), and SEP (E), maintained a 2:1 apple:Gillenia ratio (Fig. 4a). Putative stoichiometric preservation in the A/C/E floral homeotic clades is consistent with the floral quartet model of ternary complex formation for molecular function38. The predominant 2:1 ratio within these clades suggested potential for different selective pressures on duplicated genes, which was further explored with E-type MADS8 as the exemplar.
G. trifoliata as a negative control for fruit flesh development
Many successful Rosaceous fruit crops rely on the development of accessory tissues (receptacle and hypanthium) as edible or structural components. In addition, the presence of a floral hypanthium is a unifying character for all species within Rosaceae2. In apple and strawberry, genes related to Arabidopsis SEPALLATA1/2, MdMADS8/9 and FaMADS9, were implicated in the growth and development of fruit flesh39,40. Owing to intrafamilial functional similarity and importance for reproductive development, SEP1/2-like orthologues were assessed for selection pressure via the ratio of nonsynonymous/synonymous substitution rates (dN/dS, ω). A series of branch-wise hypotheses were tested using a phylogeny of Rosid SEP1/2-like genes (Fig. 5, Supplementary Table 12a). It was found that Rosaceae SEP1/2-like genes had a highly significant (p < 0.005) near twofold increase towards positive selection compared with non-Rosaceae orthologues (H1; Fig. 5a). A 3-ratio test suggested uneven selection pressure across Amygdaloideae subfamily (H2; Supplementary Fig. 23a). Therefore, a hypothesis was tested in which Rosaceae species with accessory-dependent fruit (Maleae and Rosoideae) were under different selective pressures from those without (Gillenia, Prunus and Cerasus). This hypothesis showed a clear twofold increase towards positive selection of SEP1/2-like genes in accessory-dependent fruiting Rosaceae and was statistically significant in both 3-ratio (H3; Fig. 5b) and 2-ratio (H4; Fig. 5c) hypotheses. Hypotheses testing for different selection pressures between Maleae homoeologues (MADS8 vs. MADS9), were not statistically significant (H5 & H6; Supplementary Fig. 23a). A second study was conducted for SEP1/2-like orthologues without Gillenia, and while the 2-ratio hypothesis (H2) was found to be statistically significant, the 3-ratio hypothesis was not (H3; Supplementary Table 12b; Supplementary Fig. 23b), suggesting Gillenia provided greater statistical power along with the ‘negative control’, fleshless non-accessory-dependent phenotype. Lastly, a third study was conducted for SEP3-like Rosid orthologues. Arabidopsis SEP3 is a central hub protein in floral gene networks41 but has no documented relationship with accessory flesh development. Here, the increase towards positive selection was reduced, both 2-ratio hypotheses (H1 & H3) were less significant compared to corresponding SEP1/2-like hypotheses (Fig. 5a, c), and the 3-ratio hypothesis (H2) was not statistically significant (Supplemental Table 12c; Supplementary Fig. 23c).

a Two-ratio hypothesis: Rosaceae versus non-Rosaceae orthologues. b Three-ratio hypothesis: accessory-dependent fruiting Rosaceae versus non-accessory-dependent Rosaceae versus non-Rosaceae orthologues. c Two-ratio hypothesis: accessory-dependent fruiting Rosaceae versus non-accessory-dependent fruiting Rosaceae and non-Rosaceae orthologues. Prefixes: pear (Pb, Pyrus betuleafolia; Pc, P. communis BartlettDH); apple (Mb, Malus baccata; Md, M. domestica GDDH13); Gt, Gillenia trifoliata; Pruper, Prunus persica (peach); Prudul, P. dulcis; Ceryed, Cerasus yeodonis; Pruavi, P. avium; Rubocc, Rubus occidentalis (raspberry); Roschi, Rosa chinensis; Fraves, Fragaria vesca (strawberry); Cansat, Cannabis sativa; Medtru, Medicago truncatula; Poptri, Populus trichocarpa; At, Arabidopsis thaliana; Vv, Vitis vinifera. 2Δl, Likelihood ratio test against Χ2(df = 1, a = 0.05) = 3.841
Discussion
The combination of phenotypic and genomic resources provided here will facilitate the use of G. trifoliata as a model species and ‘rosetta stone’ for translational science. The phenotype of G. trifoliata has been described in detail in a BBCH framework, to enhance comparative studies within Rosaceae, and to characterise the development of characters of interest, such as annual bearing, cane-like habit, and dry fruit development. The dry fruit phenotype provides a new tool, previously unavailable for apple and pear, to study fleshy fruit development in these two economically important fruit crops. The G. trifoliata genome provides new perspectives on genome dynamics relating to growth and reproductive development within Rosaceae. The absence of ANR1 genes in G. trifoliata, strawberry, and raspberry correlates with a herb/shrub growth habit and specifically in G. trifoliata may correlate with its reversion to a rhizomatous herb habit after separation from the Maleae ancestor42. Many clade contractions were observed in G. trifoliata compared with apple, particularly around flowering- and dormancy-related MADS genes, which may correlate with its annual bearing habit. A tandem NAC gene array, which houses a ripening-associated NAC in apple35, has expanded greatly in fleshy-fruiting apple, but appears to have contracted in dry-fruiting Gillenia relative to fleshy-fruiting strawberry, and provides additional support for further study of NAC-regulated fleshy fruit ripening. Overall, gene family analysis revealed a strong retention of a 2:1 apple:Gillenia ratio, which demonstrates preferential retention of homoeologues in these families rather than selective pressure in favour of segmental duplication and clade expansion. In contrast to this was the observation of a Maleae-conserved 1:1 ratio in an apple homologue of BLR, an important floral gene network hub gene with roles in regulating organ boundaries28 and that provides a candidate to begin unravelling the mechanism behind ovary-hypanthium fusion, which has been pivotal in the development of the pome-fruit structure. The case study of selective pressures acting upon MADS8-like genes in Rosids using the ratio of nonsynonymous to synonymous substitutions demonstrated the power of using Gillenia as a negative control for fleshy-accessory fruit development and exemplifies its potential utility for future studies of fleshy fruit evolution.
Methods
Plant materials
Mature plants of G. trifoliata were obtained from Wake Robin Nursery, Balclutha, New Zealand (https://www.wakerobin.co.nz/). During the growing season, plants were held in glasshouse conditions at Plant & Food Research, Auckland, New Zealand with automated watering and automated shading at temperatures above 28 °C. During winter, plants were held in a cold room for twelve weeks at 5 °C to simulate overwintering. Flowers were outcrossed and hand-pollinated to induce fruit set. Phenotypic assessments were performed across three multi-stemmed individuals.
Molecular extraction, library preparation and sequencing
High molecular weight (HMW) nuclear genomic DNA (ngDNA) was extracted from isolated nuclei prepared as described in43 with the following modifications: 1.5 g mature leaves (for 10X Chromium) or 2 g young leaves (for Oxford Nanopore) were fine ground in liquid nitrogen with a mortar and pestle, suspended in 300 mL nuclei isolation buffer and filtered through two layers of Miracloth. All centrifugation steps were performed at 1800 × g at 10 °C. HMW ngDNA was extracted from isolated nuclei using the CTAB/NaCl/Proteinase K method described in ref. 44 with the following modifications: nuclei pellet was re-suspended in 15 mL CTAB buffer supplemented with 2 mg Proteinase K, and after lysis incubation was extracted with an equal volume of 24:1 chloroform:isoamyl alcohol. QC was performed with Qubit dsDNA high sensitivity assay (Life Technologies), Nanodrop spectrophotometer (Thermo Fisher Scientific) and pulsed-field gel electrophoresis. HMW ngDNA was supplied to Novogene (Hong Kong) for Illumina 10X Chromium sequencing which generated short-read data of 109X coverage, and to the Australian Genome Research Facility (AGRF; Melbourne, Australia) for long-read Oxford Nanopore Technologies (ONT) sequencing which generated data of 123X coverage.
For long-range Hi-C sequencing, chromatin was extracted from isolated nuclei purified using polyvinylpolypyrrolidone and Percoll gradients as described in ref. 45. Hi-C libraries were prepared according to ref. 46 with modifications. Nuclei were crosslinked with 4% formaldehyde, quenched, washed, lysed and chromatin normalised according to Dovetail™ Hi-C kit (Dovetail Genomics). Chromatin fragmentation and biotinylation were performed with Fragmentation buffer and enzyme mix from Phase Genomics Hi-C kit for Plants v1 (Phase Genomics). Ligation was achieved with T4 DNA ligase and buffer (Invitrogen), at 16 °C overnight with rotation. Crosslink reversal was performed with 0.4 μg/μL Proteinase K (Qiagen) in 1X CutSmart buffer (New England Biolabs) and DNA purified with 2X AMPure XP Beads. The short insert library was prepared with NEBNext Ultra II FS DNA library prep kit for Illumina (New England Biolabs). DNA was fragmented for 8 min in dsDNA fragmentase enzyme mix, ligated with Illumina adaptor, digested with USER enzyme mix and purified with AMPure XP beads. Biotinylated molecules were captured with Dynabeads M280 (Invitrogen) according to the manufacturer’s protocol. The captured library was amplified with NEB Q5U DNA polymerase, the universal Illumina primer and an index primer 12 with the following PCR profile: 98 °C, 30”-(98 °C, 10”−62 °C, 30”−65 °C, 60”)x14-65 °C, 5 min- stop. The amplified library was size selected with AMPure XP beads at 0.6X/0.2X). The average fragment size of the selected amplicons was 399 bp estimated by capillary electrophoresis (Fragment Analyzer). The amplicons were sequenced by AGRF (Australia).
Short-read RNA-Seq libraries were prepared for three floral stages following47 and sequenced by AGRF (Australia) on HiSeq platform (Illumina). Long-read RNA libraries were prepared for shoot, root and leaf tissues using the Direct cDNA Sequencing kit SQK-DCS109 (Oxford Nanopore) and sequenced with Oxford Nanopore minION device using Flowcell FLO-MIN106.
Genome assembly
The genome size of G. trifoliata was estimated using flow cytometry as described in ref. 48. Leaves from Malus domestica ‘Royal Gala’ or G. trifoliata were co-chopped with each of two internal reference standards, Trifolium repens or Bellis perennis. Resulting ratios were used to deduce the estimated genome size for Gillenia using the estimated genome size for apple from the latest HFTH1 genome18 of 708.54 Mbp.
Sequencing data quality was assessed using FastQC (v0.11.7)49 and PycoQC (v2.5.0.21)50 for 10X Chromium and ONT reads, respectively. Hi-C data quality was assessed using BWA (v0.7.17)51 following the PhaseGenomics read mapping strategy (https://phasegenomics.github.io/2019/09 /19/hic-alignment-and-qc.html) and the script “hic_qc.py” (https://github.com/phasegenomics/hic_qc). K-mers (k = 21) were generated from 10X Chromium reads using Jellyfish (v2.2.10)52. The k-mer count histogram and the genome heterozygosity were analysed and visualised with the online version of GenomeScope2 (http://qb.cshl.edu/genomescope/genomescope2.0/)53. The initial de novo haplotype assemblies were generated from 10X Chromium short reads with effective coverage of 45X using Supernova (v1.2.2)54 (‘–maxreads=130716163’). The haplotype assembly with longer N50 and higher BUSCO score was selected, contaminating sequences were removed with Kraken255 against RefSeq93 database56, and allelic contigs were identified and reassigned using Purge Haplotigs57. The later-obtained ONT reads with 123X coverage were assembled using FLYE (v2.5)58 and subsequent contigs were used for post-scaffolding the cleaned 10X Chromium-based assembly using RaGOO59 followed by gap-filling with ONT reads using TGS-GapCloser (v1.1.1)60. Hi-C data were used to construct chromosomal-level assembly utilising HiC-Suite (https://github.com/pfrnz/HiC-Suite), to construct joins based on visualising Hi-C contact maps. The resultant assembly was then compared and optimised with Hi-C scaffolding results produced from ALLHIC (v0.9.8)61 to additionally join a few short scaffolds. Results from a syntenic map with the Malus domestica genome18 generated from MUMmer (v4.0.0)62 were used to further inform some Hi-C interactions missed in previous steps. Final pseudo-chromosomes were determined with a whole-genome Hi-C contact map. Duplicated un-anchored sequences were removed using ‘gt sequniq’ in GenomeTools63. Genome assembly completeness was assessed by mapping 10X Chromium Illumina short reads to the assembly using bowtie2 (2.3.4.3)64, and with BUSCO analyses (v3.0.2)16 against “embryophyta_odb10” database (updated on 2019-11-21).
Genome annotation
TEs were detected with EDTA pipeline (v1.9.3)65 using parameters “–species others –step all –sensitive 1 –anno 1 –evaluate 1 –threads 20”. The Gillenia genome was further soft-masked with RepeatMasker-open-4-0-5 (-pa 20 -e ncbi -gff -xsmall –poly). Illumina short-read RNA-seq data from three flower stages were mapped to the soft-masked genome using STAR (v2.6.1d)66 (–readFilesCommand zcat –outSAMattrRGline ID:$sample –runThreadN $thread –outBAMsortingThreadN $thread –outSAMtype BAM SortedByCoordinate). ONT long-read RNA-seq data were called with guppy/4.2.2. Adapters were removed and chimeric reads were split with porechop/0.2.3, then further filtered with quality (Q7) and minimum read length of 300 using filtlong v0.2. The cleaned reads were mapped to the Gillenia genome with minimap2/2.17 -ax splice. Alignments were compressed and indexed with samtools v1.10. Quality checks on ONT sequencing data, long RNA reads, and mapping were carried out with MinIONQC.R, FastQC v0.11.7, and qualimap v2.2.1, respectively. Taxonomy classification of the cleaned ONT RNA-seq reads was performed with Kraken255 against RefSeq93 database56, and visualised with Krona (v2.7). The ONT long-read and Illumina short-read RNA-seq alignments were used as hints in BRAKER (v2.1.0)67 to inform gene prediction with parameters “–species = Gtr.NCBI.softmasking –gff3 –cores = $thread –workingdir = $outDir –augustus_args = ‘–strand = both –genemodel = complete –alternatives-from-evidence = true –noInFrameStop = true’ –overwrite”. Transfer RNA was detected with tRNAscan-SE (v2.0)68.
Genome synteny and ancestral reconstruction
Dot plots were generated using several commands from MUMmer (v4.0.0 beta2)62. Syntenic blocks were produced using “nucmer –mum”, then “delta-filter” with appropriate nucleotide identity (-i) and syntenic block length (-l) settings for every comparison, and finally “mummerplot” with the given options of “–large –png -R -Q –filter –layout -p”. Dots presented for Gillenia versus Malus18 and Pyrus19 chromosomes have 70% nucleotide identity with a minimum length cutoff of 5 kb. Dots presented for Gillenia versus Prunus17 and Rubus20 have 70% nucleotide identity with a minimum length cutoff of 2 and 1 kb respectively. To construct the ancestral genome, forty-four syntenic blocks were identified across the dot plots between Gillenia versus Malus, Prunus, and Rubus. Blocks were numbered and owing to high syntenic conservation were able to be manually assembled to elucidate most parsimonious genomic rearrangements. Ancestral blocks in descendent genomes were drawn in MapChart69 and ancestral chromosome sizes were estimated based on size of descendant blocks.
The circos plot of genomic synteny between apple and G. trifoliata was generated using Circos visualisation tool (v0.69-6)70. The homological regions were produced in advance using nucmer (–mum) and dnadiff with default options from MUMmer62. Multiple-to-multiple coordinates file “mcoords” filtered out regions shorter than 100 bp length and then reformatted and inputted in the command “bundlelinks” (-max_gap 10000 -min_bundle_size 10000 -min_bundle_membership 3) from circos tools (v0.22) within Circos package to build connecting lines. The final plot was generated using “circos” command with customised config file from Circos.
Comparative transposable element identification
Genome assemblies were collected from rosaceae.org, including Malus domestica (GDDH13 and HFTH1), Pyrus communis (BartlettDHv2), P. betulaefolia (PbeSD), P. persica v2.0, Rubus occidentalis v2.1, and F. vesca v4.0.a1. EDTA (v1.9.3)65, a filtering package based on multiple repeats detection tools such as LTR_Finder, LTRharvest, HelitronScanner, LTR_retriever, TIR-Learner and RepeatModeler, was run in the conda environment in a consistent way on the Gillenia genome and downloaded genome assemblies. The cross-species summary was drawn based on the result in EDTA.intact.gff3 and EDTA.TEanno.gff3 files for each genome.
Orthologous gene clustering
OrthoVenn271 was used to identify orthologous gene clusters between Gillenia and selected species of Rosaceae. Predicted protein lists were obtained from Genome Database for Rosaceae (rosaceae.org) for F. vesca v4.0.a272, R. occidentalis v3.020, P. persica v2.0.a117, M. domestica GDDH13 v1.122, and P. betulaefolia v1.032.
Gene family identification and phylogenetic analysis
Protein sequences identified using the first apple genome5 and Arabidopsis were used to perform all-to-all BLASTp searches (e-value 1e−50) against predicted protein models from two recent apple genomes, GDDH13 and HFTH118,22 and the Gillenia genome. GDDH13 gene models were used in preference to HFTH1 models as the base set because there was a higher incidence of concatenated gene models in the HFTH1 predictions. Reciprocal BLASTn was used to link apple models from the same locus in the two apple genomes and to identify unique models present only in one genome. Complete lists of Gillenia and Malus models were then used to perform BLASTp searches (e-value 1e−05) of each other’s genomes. To validate familial relationships, all models were subject to MEME analysis73 to confirm the presence of familial motifs and were also checked against the NCBI Conserved Domains Database (CDD). Protein sequences were aligned in Geneious 10.2.6 (Biomatters Ltd, Auckland, NZ) using ClustalW and BLOSUM cost matrix. For each alignment, the region of significant alignment was isolated and used to generate a phylogenetic tree with PhyML74 using the JTT substitution model and bootstrap analysis of 100 data-sets. For clade-specific analyses of additional Rosaceae species, the following annotations were used: P. communis BartlettDH19, P. betulaefolia32, P. persica v2.0.a117, F. vesca v4.0.a272, R. occidentalis v3.020, and Rosa chinensis ‘Old Blush’ v275.
dN/dS analysis
Orthologues to Gillenia SEP1/2-like Gtr03.g9252.t1 and SEP3-like Gtr04.g13187.t2 were identified by reciprocal best BLAST hits. Rosaceae species, M. domestica GDDH1322, M. baccata v131, P. communis BartlettDH19, P. betulaefolia32, P. persica v2.0.a117, P. dulcis v176, P. avium v177, Cerasus x yedoensis v178, F. vesca v4.0.a272, R. occidentalis v3.020, and R. chinensis ‘Old Blush’ v275 were found using Genome Database for Rosaceae (https://www.rosaceae.org/) BLAST tools. Non-Rosaceae species, Cannabis sativa, Medicago truncatula, Populus trichocarpa, and Vitis vinifera were found using NCBI (https://www.ncbi.nlm.nih.gov/) BLAST tools, and Arabidopsis thaliana was found using TAIR (arabidopsis.org) BLAST tools. For Rosaceae species orthologues were also confirmed by synteny where possible. Protein and nucleotide alignments were performed with Clustal Omega (v 1.2.4)79 using default parameters and nucleotide alignments were manually corrected to ensure codon-alignment. Branch-wise dN/dS ratios were calculated with codeml in PAML v4.9i80.
Data availability
The data have been deposited at DDBJ/ENA/GenBank under the accession JAEHOF000000000, BioProject PRJNA669900, BioSample SAMN16480607. The Gillenia genome is available in the Genomic Database for Rosaceae (GDR): www.rosaceae.org.
References
Liu, Z., Ma, H., Jung, S., Main, D. & Guo, L. Developmental mechanisms of fleshy fruit diversity in Rosaceae. Annu. Rev. Plant Biol. 71, 547–573 (2020).
Potter, D. et al. Phylogeny and classification of Rosaceae. Plant Syst. Evol. 266, 5–43 (2007).
Alioto, T. et al. Transposons played a major role in the diversification between the closely related almond and peach genomes: results from the almond genome sequence. Plant J. 101, 455–472 (2020).
Buti, M. et al. The genome sequence and transcriptome of Potentilla micrantha and their comparison to Fragaria vesca (the woodland strawberry). Gigascience 7, 1–14 (2018).
Velasco, R. et al. The genome of the domesticated apple (Malus x domestica Borkh.). Nat. Genet. 42, 833–839 (2010).
Campbell, C. S., Evans, R. C., Morgan, D. R., Dickinson, T. A. & Arsenault, M. P. Phylogeny of subtribe Pyrinae (formerly the Maloideae, Rosaceae): limited resolution of a complex evolutionary history. Plant Syst. Evol. 266, 119–145 (2007).
Evans, R. C. & Campbell, C. S. The origin of the apple subfamily (Maloideae; Rosaceae) is clarified by DNA sequence data from duplicated GBSSI genes. Am. J. Bot. 89, 1478–1484 (2002).
Vilanova, S., Sargent, D. J., Arús, P. & Monfort, A. Synteny conservation between two distantly-related Rosaceae genomes: Prunus (the stone fruits) and Fragaria (the strawberry). Bmc Plant Biol. 8, 8 (2008).
Illa, E. et al. Comparative analysis of rosaceous genomes and the reconstruction of a putative ancestral genome for the family. Bmc Evolut. Biol. 11, 11 (2011).
Jung, S. et al. Whole genome comparisons of Fragaria, Prunus and Malus reveal different modes of evolution between Rosaceous subfamilies. Bmc Genomics 13, 13 (2012).
Zhang, Q. et al. The genome of Prunus mume. Nat. Commun. 3, 1318 (2012).
Griesmann, M. et al. Phylogenomics reveals multiple losses of nitrogen-fixing root nodule symbiosis. Science 361, 6398 (2018).
Meier, U. et al. The BBCH system to coding the phenological growth stages of plants – history and publications. J. f.ür. Kulturpflanzen 61, 41–52 (2009).
Meier, U. E. Growth stages of mono- and dicotyledonous plants: BBCH monograph. (Federal Biological Research Centre for Agriculture and Forestry, 2001).
Boyes, D. C. et al. Growth stage-based phenotypic analysis of Arabidopsis: a model for high throughput functional genomics in plants. Plant Cell 13, 1499–1510 (2001).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Verde, I. et al. The Peach v2.0 release: high-resolution linkage mapping and deep resequencing improve chromosome-scale assembly and contiguity. BMC Genomics 18, 225 (2017).
Zhang, L. et al. A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat. Commun. 10, 1494 (2019).
Linsmith, G. et al. Pseudo-chromosome-length genome assembly of a double haploid “Bartlett” pear (Pyrus communis L.). Gigascience 8, 12 (2019).
VanBuren, R. et al. A near complete, chromosome-scale assembly of the black raspberry (Rubus occidentalis) genome. Gigascience 7, 8 (2018).
Barker, M.S., Baute, G. J. & Liu, S-.L. in Plant Genome Diversity (ed. Wendel, J. F.) (Springer-Verlag, Vienna, 2012).
Daccord, N. et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 49, 1099–1106 (2017).
Parenicová, L. et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell 15, 1538–1551 (2003).
Alvarez-Buylla, E. R. et al. MADS-box genes underground becoming mainstream: plant root developmental mechanisms. N. Phytol. 223, 1143–1158 (2019).
Guitton, B. et al. Genetic control of biennial bearing in apple. J. Exp. Bot. 63, 131–149 (2012).
Wu, R. et al. SVP-like MADS box genes control dormancy and budbreak in apple. Front. Plant Sci. 8, 477 (2017).
Kumar, G. et al. Comparative phylogenetic analysis and transcriptional profiling of MADS-box gene family identified DAM and FLC-like genes in apple (Malusx domestica). Sci. Rep. 6, 20695 (2016).
Chen, D., Yan, W., Fu, L. Y. & Kaufmann, K. Architecture of gene regulatory networks controlling flower development in Arabidopsis thaliana. Nat. Commun. 9, 4534 (2018).
Li, Y. et al. Transcriptomic analysis reveals the regulatory module of apple (Malus x domestica) floral transition in response to 6-BA. BMC Plant Biol. 19, 93 (2019).
Li, R. et al. Genomewide analysis of homeobox gene family in apple (Malus domestica Borkh.) and their response to abiotic stress. J. Genet. 98, 98 (2019).
Chen, X. et al. Sequencing of a wild apple (Malus baccata) genome unravels the differences between cultivated and wild apple species regarding disease resistance and cold tolerance. G3 (Bethesda) 9, 2051–2060 (2019).
Dong, X. et al. De novo assembly of a wild pear (Pyrus betuleafolia) genome. Plant Biotechnol. J. 18, 581–595 (2020).
Kamiuchi, Y., Yamamoto, K., Furutani, M., Tasaka, M. & Aida, M. The CUC1 and CUC2 genes promote carpel margin meristem formation during Arabidopsis gynoecium development. Front. Plant Sci. 5, 165 (2014).
Moyano, E. et al. Genome-wide analysis of the NAC transcription factor family and their expression during the development and ripening of the Fragaria x ananassa fruits. PLoS ONE 13, e0196953 (2018).
Busatto, N., Tadiello, A., Trainotti, L. & Costa, F. Climacteric ripening of apple fruit is regulated by transcriptional circuits stimulated by cross-talks between ethylene and auxin. Plant Signal Behav. 12, e1268312 (2017).
Chagné, D. et al. The draft genome sequence of European pear (Pyrus communis L. ‘Bartlett’). PLoS ONE 9, e92644 (2014).
Pilkington, S. M. et al. A manually annotated Actinidia chinensis var. chinensis (kiwifruit) genome highlights the challenges associated with draft genomes and gene prediction in plants. BMC Genomics 19, 257 (2018).
Theissen, G. & Saedler, H. Plant biology. Floral quartets. Plant Biol. Flor. Quartets Nat. 409, 469–471 (2001).
Ireland, H. S. et al. Apple SEPALLATA1/2-like genes control fruit flesh development and ripening. Plant J. 73, 1044–1056 (2013).
Seymour, G. B. et al. A SEPALLATA gene is involved in the development and ripening of strawberry (Fragaria x ananassa Duch.) fruit, a non-climacteric tissue. J. Exp. Bot. 62, 1179–1188 (2011).
Rumpler, F., Theissen, G. & Melzer, R. A conserved leucine zipper-like motif accounts for strong tetramerization capabilities of SEPALLATA-like MADS-domain transcription factors. J. Exp. Bot. 69, 1943–1954 (2018).
Xiang, Y. et al. Evolution of Rosaceae fruit types based on nuclear phylogeny in the context of geological times and genome duplication. Mol. Biol. Evol. 34, 262–281 (2017).
Naim, F. et al. Advanced engineering of lipid metabolism in Nicotiana benthamiana using a draft genome and the V2 viral silencing-suppressor protein. PLoS ONE 7, e52717 (2012).
Hilario, E. Plant nuclear genomic DNA preps. protocols.io (2018).
Hilario, E. Plant nuclei enrichment for chromatin capture-based Hi-C library protocols. Protocols.io (2019).
McCartney, A. et al. An exploration of assembly strategies and quality metrics on the accuracy of the Knightia excelsa (rewarewa) genome. bioRxiv 2020.10.28.358903 (2020).
Zhong, S. et al. High-throughput illumina strand-specific RNA sequencing library preparation. Cold Spring Harb. Protoc. 2011, 940–949 (2011).
Chagné, D. et al. Polyploid and aneuploid detection in apple using a single nucleotide polymorphism array. Tree Genet. Genomes 11, 5 (2015).
Andrews, S, FastQC: a quality control tool for high throughput sequence data www.bioinformatics.babraham.ac.uk/projects/fastqc. (Babraham Bioinformatics, Babraham Institute, Cambridge, UK, 2010).
Leger, A. & Leonardi, T. pycoQC, interactive quality control for Oxford Nanopore Sequencing. J. Open Source Softw. 4, 1236 (2019).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Weisenfeld, N. I., Kumar, V., Shah, P., Church, D. M. & Jaffe, D. B. Direct determination of diploid genome sequences. Genome Res. 27, 757–767 (2017).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
O'leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinforma. 19, 460 (2018).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Alonge, M. et al. RaGOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 20, 224 (2019).
Xu, M. et al. TGS-GapCloser: a fast and accurate gap closer for large genomes with low coverage of error-prone long reads. Gigascience 9, giaa094 (2020).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845 (2019).
Marçais, G. et al. MUMmer4: a fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944 (2018).
Gremme, G., Steinbiss, S. & Kurtz, S. GenomeTools: a comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans. Comput. Biol. Bioinform 10, 645–656 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform 3, lqaa108 (2021).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol. Biol. 1962, 1–14 (2019).
Voorrips, R. E. MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 93, 77–78 (2002).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Xu, L. et al. OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 47, W52–W58 (2019).
Li, Y., Pi, M., Gao, Q., Liu, Z. & Kang, C. Updated annotation of the wild strawberry Fragaria vesca V4 genome. Hortic. Res. 6, 61 (2019).
Bailey, T. L. & Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int Conf. Intell. Syst. Mol. Biol. 2, 28–36 (1994).
Guindon, S. & Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003).
Raymond, O. et al. The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 50, 772–777 (2018).
Sánchez-Pérez, R. et al. Mutation of a bHLH transcription factor allowed almond domestication. Science 364, 1095–1098 (2019).
Wang, J. et al. A de novo assembly of the sweet cherry (Prunus avium cv. Tieton) genome using linked-read sequencing technology. PeerJ 8, e9114 (2020).
Shirasawa, K. et al. Phased genome sequence of an interspecific hybrid flowering cherry, ‘Somei-Yoshino’ (Cerasus x yedoensis). DNA Res. 26, 379–389 (2019).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Zhu, T., Nevo, E., Sun, D. & Peng, J. Phylogenetic analyses unravel the evolutionary history of NAC proteins in plants. Evolution 66, 1833–1848 (2012).
Ooka, H. et al. Comprehensive analysis of NAC family genes in Oryza sativa and Arabidopsis thaliana. DNA Res. 10, 239–247 (2003).
Acknowledgements
This study was supported by the New Zealand Ministry for Business, Innovation, and Employment (MBIE) Strategic Science Investment Fund (SSIF) platform ‘Genomics Aotearoa’ (www.genomics-aotearoa.org.nz), and SSIF funding allocated to Plant & Food Research (PFR) and Technology Development funding from PFR aligned with pipfruit research.
Author information
Authors and Affiliations
Contributions
H.I. grew the plants, performed the phenological analysis, analysed gene families, performed dN/dS analysis, and wrote the manuscript. C.W., C.D. and R.C. performed the bioinformatics analysis, including genome assembly and annotations. E.H. and H.I. performed lab experiments. D.C. and C.W. did the synteny analysis and reconstructed the ancestral genome of Rosaceae. A.S. did the Hi-C scaffolding. S.E. did the flow cytometry analysis. H.I., R.S., K.D., C.D. and D.C. designed the project. All authors approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ireland, H.S., Wu, C., Deng, C.H. et al. The Gillenia trifoliata genome reveals dynamics correlated with growth and reproduction in Rosaceae. Hortic Res 8, 233 (2021). https://doi.org/10.1038/s41438-021-00662-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-021-00662-4
