
Abstract
Morinda officinalis is a well-known medicinal and edible plant that is widely cultivated in the Lingnan region of southern China. Its dried roots (called bajitian in traditional Chinese medicine) are broadly used to treat various diseases, such as impotence and rheumatism. Here, we report a high-quality chromosome-scale genome assembly of M. officinalis using Nanopore single-molecule sequencing and Hi-C technology. The assembled genome size was 484.85 Mb with a scaffold N50 of 40.97 Mb, and 90.77% of the assembled sequences were anchored on eleven pseudochromosomes. The genome includes 27,698 protein-coding genes, and most of the assemblies are repetitive sequences. Genome evolution analysis revealed that M. officinalis underwent core eudicot γ genome triplication events but no recent whole-genome duplication (WGD). Likewise, comparative genomic analysis showed no large-scale structural variation after species divergence between M. officinalis and Coffea canephora. Moreover, gene family analysis indicated that gene families associated with plant–pathogen interactions and sugar metabolism were significantly expanded in M. officinalis. Furthermore, we identified many candidate genes involved in the biosynthesis of major active components such as anthraquinones, iridoids and polysaccharides. In addition, we also found that the DHQS, GGPPS, TPS-Clin, TPS04, sacA, and UGDH gene families—which include the critical genes for active component biosynthesis—were expanded in M. officinalis. This study provides a valuable resource for understanding M. officinalis genome evolution and active component biosynthesis. This work will facilitate genetic improvement and molecular breeding of this commercially important plant.
Similar content being viewed by others

Introduction
Morinda officinalis How, belonging to the genus Morinda of the family Rubiaceae, is a perennial vine naturally distributed in southern China and northern Vietnam (Fig. 1a)1. M. officinalis, a commonly used traditional Chinese medicinal plant, was first reported in Shen Nong Ben Cao Jing and accepted in the Chinese Pharmacopoeia in 1963. The roots of M. officinalis, named bajitian in traditional Chinese medicine, are one of the four famous southern herbs from the Lingnan region of southern China. The roots of M. officinalis are widely used for the treatment of various diseases, such as impotence, infertility, abnormal menstruation, rheumatism, and arthralgia (Fig. 1b, c)2,3. In China and northeast Asia, M. officinalis is also usually used as a tonic for nourishing the kidneys and enhancing immune functioning in the body2,4.

a The M. officinalis plant. b The roots of M. officinalis with the woody core removed (named bajitian in traditional Chinese medicine). c The roots of M. officinalis at different growth stages
Phytochemical studies have shown that M. officinalis contains anthraquinones, iridoids, flavonoids, polysaccharides, volatile oils, and other important compounds5,6. Anthraquinones are one of the main active components and mainly contain physcion, rubiadin-1-methylether, anthragallol-2-methylether, etc., which have various biological activities such as antibacterial, anticancer, anticoagulant, and antiviral activities7,8. Monotropein, a kind of iridoid compound, has potent antiinflammatory and analgesic effects and is the main component of M. officinalis for dispelling wind and eliminating dampness9. The polysaccharides in M. officinalis mainly consist of glucose and fructose, which have antifatigue, antidepressant, and antiosteoporosis roles10,11. Various pharmacological and clinical studies have linked M. officinalis with aphrodisiac and immunomodulatory effects and antiosteoporosis, antidepression and antiinflammatory properties12,13,14.
In China, Guangdong Province is the main planting area for M. officinalis, accounting for approximately 90% of its output. In this province, Gaoyao District and Deqing County (Zhaoqing City) are authentic M. officinalis-producing areas15,16. In recent years, wild resources of M. officinalis have been significantly threatened in China to a level risking extinction due to a sharp increase in M. officinalis market demand, especially in Guangdong Province15. Therefore, artificially cultivated M. officinalis has become the primary source of related medicinal materials in China. M. officinalis has a long cultivation period of approximately five years; moreover, its sexual reproduction cycle is complex. Long-term asexual reproduction of M. officinalis has led to germplasm depletion that seriously affects its quality and yield. Thus, diseases of M. officinalis are becoming a severe issue, especially stem rot caused by Fusarium oxysporum, which is devastating17. Thus, the breeding of promising new varieties is urgently needed.
The genomic information of M. officinalis can lay the foundation for improving the quality of these medicinal materials, accelerating molecular breeding, protecting wild resources, and aiding the discovery and utilization of functional genes18. Whole-genome sequencing has been performed for many medicinal plants, such as Scutellaria baicalensis, Isatis indigotica, weeping forsythia, and Macleaya cordata19,20,21,22. However, the genome sequence of M. officinalis has not yet been reported, which restricts the development of functional genomics and molecular breeding of this plant. In this study, we generated a high-quality genome for M. officinalis by nanopore sequencing and Hi-C technology to elucidate its genomic characteristics. Transcriptome sequencing was also carried out to identify the candidate genes related to active compound biosynthesis. The reference genome information obtained in this research will be a valuable resource for promoting genetic improvement and understanding the biosynthesis of active ingredients of this medicinal plant.
Results
Genome assembly and quality validation
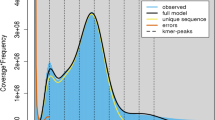
To evaluate the genome size and heterozygosity of M. officinalis, a total of 61.4 Gb short reads from the MGISEQ-2000 sequencing platform were subjected to K-mer analysis (Supplementary Table S1). The 17-mer frequency curve showed a bimodal distribution, with the highest peak occurring at a depth of 54 (Supplementary Fig. S1a). Based on the total number of K-mers, the genome size and heterozygosity of M. officinalis were estimated to be 485.4 Mb and 1.32%, respectively (Supplementary Fig. S1b). These results indicated that the genome of M. officinalis was small but highly heterozygous.
De novo assembly of the 62.92 Gb of single-molecule long reads from the Oxford Nanopore PromethION sequencing platform was performed with NextDenovo software (Supplementary Table S1). After removing redundant and contaminated sequences (nontarget classes, mitochondria, and chloroplasts), the final postcorrection genome size was 484.85 Mb, with a contig N50 of 4.21 Mb (Table 1). The genome size was similar to that estimated by the genome survey. Employing Hi-C technology, 398.8 million clean reads from the Illumina NovaSeq 6000 sequencing platform were used for chromosome construction to further refine the M. officinalis genome assembly. Moreover, using the agglomerative hierarchical clustering method in LACHESIS software, a total of 99.94% of the assembly was anchored to 11 pseudochromosomes; the size ranged from 33.06 Mb to 47.00 Mb with a contig N50 of 3.61 Mb and scaffold N50 of 40.97 Mb (Table 1 and Supplementary Table S2). Finally, the contig sequences were connected in the determined order and direction by adding 100 N to obtain the final chromosome-level genome sequence with a chromosome mount rate of 90.77% (Fig. 2 and Supplementary Table S2). A Hi-C interaction heatmap showed that the clustering, ordering, and orientation of the contigs was valid, providing the first high-quality chromosome-scale genome assembly for M. officinalis (Supplementary Fig. S2).

The circle from outside to inside represents chromosomes (a), gene density (b), tandem repeat density (c), LTR-Copia density (d), LTR-Gypsy density (e), transposon element density (f), and gene collinearity connected by curved lines (g). All distributions are drawn in a window size of 1 Mb
A variety of methods have been employed to evaluate the assembly quality of genomes. Here, the assembly completeness was evaluated using Benchmarking Universal Single-Copy Orthologs (BUSCOs). The BUSCO analysis identified 97.02% of the complete BUSCOs in the assembly (Supplementary Table S3). To assess assembly consistency, the short reads were mapped to the genome using bwa software. In total, ~99.26% of the clean data were mapped to the genome assembly (Supplementary Table S4). The mapping ratio of the RNA-seq reads from different tissues was in the range of ~93.94–96.08% (Supplementary Table S5). We further evaluated potential contamination in the genome by using GC depth analysis. The GC depth scatter plot showed that the GC content was distributed at 30–40%, and the sequencing depth was concentrated at 110–150×, indicating high quality without contamination in the data (Supplementary Fig. S3). All these results indicated high completeness and consistency of the M. officinalis genome assembly.
Genome annotation
Homology-based annotation and a de novo approach were applied to identify transposable elements (TEs) and tandem repeats in the M. officinalis genome. In total, we identified 281.41 Mb of nonredundant repetitive sequences, accounting for 58.04% of the assembled genome (Supplementary Table S6). Of these predicted repeats, TEs comprised the largest proportion (54.18%), including 42.92% Class I repeats and 11.26% Class II repeats. Long terminal repeat (LTR) retrotransposons accounted for 35.79% of the genome (Supplementary Table S6). Based on the assembled genome, a total of 209 rRNAs, 207 snRNAs, 78 miRNAs, and 644 tRNAs were predicted (Supplementary Table S7).
A total of 27,102 nonredundant protein-coding genes were predicted by using a combination of de novo, homolog-based, and transcriptome-based predictions (Table 2). The average gene length and coding sequence size were 3762.35 and 1169.11 bp, respectively, with an average of five exons per gene (Table 2). Then, we performed functional annotation of predicted genes by comparison with different databases. Overall, 24,769 (91.39%) genes were functionally annotated in at least one of the public databases, and 6153 (22.70%) genes could be annotated in all databases (Supplementary Fig. S4a). A total of 24,637 (90.90%) genes showed homologous genes in the NR database, while 20,712 (76.42%) genes were similar to proteins in the SwissProt database. Among the blastx top hits, species of the genus Coffea (Rubiaceae family) showed the highest proportion (82.77%) of homologous genes, including Coffea arabica (49.54%), Coffea eugenioides (20.44%), and C. canephora (12.79%) (Supplementary Fig. S4b). In addition, 14,998 (55.34%) genes were assigned to at least one GO term and classified into 41 GO functional subcategories (Supplementary Fig. S4c). To further understand the metabolic pathways of M. officinalis, 9737 (35.93%) genes were annotated in the KEGG pathway database (Supplementary Fig. S4d). Pathways associated with “biosynthesis of other secondary metabolites” (466 genes) and “metabolism of terpenoids and polyketides” (216 genes) can be used to explore the biosynthesis pathways of active ingredients in M. officinalis.
We conducted BUSCO analysis to verify the predicted genes. The results showed that approximately 96.80% of the complete BUSCOs could be identified in the annotated results, indicating the high reliability of the predicted results (Supplementary Table S8). The number of predicted genes and structural characteristics of the M. officinalis genome were consistent with those of related species, which indicated that the annotation results were acceptable (Supplementary Table S9).
Gene families and phylogenetic relationships
OrthoMCL software was used to identify the gene families of M. officinalis and nine other species, among which C. canephora is the most closely related species to M. officinalis and also belongs to the Rubiaceae family. A total of 22,750 (83.94%) genes were categorized into 14,124 gene families, 849 of which were unique to M. officinalis (Supplementary Table S10). In each species, 1576 genes were identified as single-copy orthologs. A total of 7230 genes in M. officinalis did not cluster with the genes of other species, indicating that these genes were M. officinalis specific (Supplementary Fig. S5a, b). Furthermore, we performed KEGG enrichment analysis to explore the metabolic pathways involved in these species-specific genes. Interestingly, we found that the pathways related to the synthesis of secondary metabolites were significantly enriched (q value < 0.05), including “indole alkaloid biosynthesis” (36 genes), “phenylpropanoid biosynthesis” (103 genes), and “stilbenoid, diarylheptanoid, and gingerol biosynthesis” (29 genes) (Supplementary Table S11). Moreover, we also identified 149 genes involved in “plant–pathogen interaction”. These data will help to reveal the molecular mechanisms underlying the interactions between M. officinalis and pathogens.
We used MAFFT software to perform multiple sequence alignments on the identified single-copy orthologous genes. The phylogenetic tree was constructed using the PROTGAMMAAUTO model of RAXML software, in which V. vinifera and Arabidopsis thaliana were outgroup species. As shown in Fig. 3a, M. officinalis was most closely related to C. canephora, and the divergence time of the two was 49.27 (31.55–65.27) million years ago (Mya). Comparative genomic analysis showed that 732 expanded and 308 contracted gene families were discovered in M. officinalis (Fig. 3b). Compared with the closely related species of C. canephora (383 expansion/370 contraction), M. officinalis showed more gene family expansion than contraction. KEGG enrichment analysis of the expanded genes suggested that they were mainly enriched in “ABC transporters” (30 genes), “starch and sucrose metabolism” (78 genes), “phenylpropanoid biosynthesis” (97 genes), “isoquinoline alkaloid biosynthesis” (19 genes) and so on, indicating that some of these might be related to the biosynthesis of active compounds (Supplementary Table S12). The contracted gene families were involved in “ether lipid metabolism” (8 genes), “endocytosis” (20 genes), “sesquiterpenoid and triterpenoid biosynthesis” (5 genes), “spliceosome” (18 genes), “protein processing in endoplasmic reticulum” (18 genes) and “plant–pathogen interaction” (25 genes), indicating that some of these families may be related to environmental adaptation (Supplementary Table S13). Notably, we found that a series of genes related to secondary metabolism and environmental adaptation exhibited significant expansion (Supplementary Table S14). These results provide valuable resources for understanding the biosynthesis of active ingredients and the interaction between M. officinalis and its growth environment.

a Phylogenetic tree and divergence time estimation. The value on each node represents the divergence time of each species. The red dots depict the fossil record used to correct the divergence time. b Gene family expansion (green) and contraction (red). c Ks distribution among M. officinalis and three other species. Lines show Ks distribution within (continuous) and between genomes (dashed). d Collinear relationship of M. officinalis, C. canephora and Vitis vinifera. The gray line connects matched gene pairs
Positive selection, WGD, and collinearity
The Ka/Ks ratios of the single-copy genes were used to evaluate the positive selection of genes in M. officinalis. A total of 101 candidate genes were strictly positively selected (p-value < 0.05) (Supplementary Table S15). GO enrichment analysis showed that these genes were enriched in “DNA repair”, “ATP-dependent helicase activity”, “chromosome”, and “DNA topological change”, indicating that these positively selected genes may improve DNA damage resistance in adverse environments. To estimate the potential WGD events of M. officinalis, synonymous nucleotide substitutions (Ks) were characterized in M. officinalis, C. canephora, Catharanthus roseus, and V. vinifera. Based on the Ks distribution between M. officinalis and the other three genomes, M. officinalis–C. canephora (0.608) divergence time was slower than that of M. officinalis–C. roseus (0.867) and M. officinalis–V. vinifera (1.238), which was consistent with the phylogenetic tree (Fig. 3c). The distribution of Ks showed one peak at ~2.1 to 2.3 in the genomes of M. officinalis, C. canephora, and V. vinifera. The results showed that they shared an ancient WGD event (the core eudicot γ triplication event) before their divergence and no recent independent WGD event (Fig. 3c). The comparative genome structure between M. officinalis and C. canephora showed high collinearity, indicating that there was no large-scale structural variation after the divergence between M. officinalis and C. canephora (Fig. 3d and Supplementary Fig. S6). For most collinear regions, one chromosome of M. officinalis corresponded to one chromosome of C. canephora; for example, MoChr2, MoChr5, MoChr6, MoChr7, MoChr8, MoChr9, and MoChr10 of M. officinalis corresponded to CcChr2, CcChr3, CcChr10, CcChr7, CcChr8, CcChr5, CcChr11 and CcChr4 of C. canephora, respectively. MoChr1 corresponded to CcChr1, CcChr2 and CcChr6; MoChr3 corresponded to CcChr2, CcChr6 and CcChr9; MoChr4 corresponded to CcChr1, CcChr3, and CcChr6 (Supplementary Fig. S6). These observations indicated that MoChr1, MoChr3, and MoChr4 might have formed by fragmentation and recombination of ancestral chromosomes. We further conducted intergenomic collinearity between M. officinalis and V. vinifera. The M. officinalis genome generally showed a one-to-one syntenic relationship with V. vinifera, which was consistent with the result that the M. officinalis genome did not undergo a recent WGD event (Fig. 3d). Interestingly, the collinear regions of MoChr5 mainly corresponded to VvChr5, and MoChr6 corresponded to VvChr 18, while other chromosomes, especially MoChr1, MoChr2, and MoChr3, did not exhibit any significant corresponding relationships between M. officinalis and V. vinifera (Supplementary Fig. S6).
Characteristic analysis of genes showing organ-specific expression
Based on gene expression levels, we identified 451, 254, 109, 165, and 219 genes expressed explicitly in stalks, leaves, one-year-old roots (AR), three-year-old roots (TR), and six-year-old roots (SR), respectively, and 17,578 genes were expressed in all tissues (Fig. 4a). To elucidate the similarities and differences of gene expression patterns in different tissues, we also performed a k-means cluster analysis. A total of 16,771 differentially expressed genes (DEGs) were divided into 10 clusters (Fig. 4b). We, therefore, focused our attention on the clusters that contained genes with tissue-specific expression. We found that the leaf-biased genes (cluster 1) were highly correlated with fundamental pathways, such as “photosynthesis”, “biosynthesis of secondary metabolites”, “photosynthesis-antenna proteins”, “carbon fixation in photosynthetic organisms” and “porphyrin and chlorophyll metabolism” (Supplementary Table S16). In contrast, stalk-biased genes (cluster 9) were significantly associated with defense responses, such as “MAPK signaling pathway-plant” and “plant–pathogen interaction” (Fig. 4c and Supplementary Table S16). Additionally, we also identified that genes in clusters 2, 4, and 8 were highly expressed at different developmental stages of roots, and some genes were continuously enhanced with root development (clusters 3 and 7). The functions of these genes were mainly enriched in “phenylpropanoid biosynthesis”, “amino sugar and nucleotide sugar metabolism”, “ABC transporters”, “MAPK signaling pathway-plant”, “plant–pathogen interaction” and “peroxisome”, indicating that they may be related to the synthesis, transport, and storage of active ingredients and defense responses (Supplementary Table S16). These results provide a basis for further analysis to reveal the gene expression regulatory network and regulate bioactive metabolite derivative production in M. officinalis.

a Venn diagram showing the expressed genes in roots, stalks, and leaves (FPKM > 0.1). The overlapping regions represent genes expressed in at least two tissues, while the nonoverlapping regions represent tissue-specific genes. b Hierarchical clustering showing the expression patterns of DEGs. The x axis represents different tissues, and the y axis represents the standardized gene expression level based on the log2 (FPKM + 1) values. AR, one-year-old roots; TR, three-year-old roots; SR, six-year-old roots. c Part of the MAPK signaling pathway. Various color blocks represent the normalized gene expression levels of the DEGs in different tissues
Identification of genes related to the anthraquinone biosynthesis pathway
Anthraquinones in Rubiaceae plants are mainly synthesized through the shikimate/o-succinylbenzoic acid pathway, which mainly involves the shikimate pathway, TCA cycle, mevalonate (MVA) pathway, and methylerythritol phosphate (MEP) pathway (Fig. 5a). 1,4-Dihydroxy-2-naphthoate (DHNA) is formed by isochorismate and α-ketoglutarate under the catalysis of a series of enzymes. Therefore, DHNA combines with dimethylallyl pyrophosphate (DMAPP), derived from the MVA and MEP pathways, to form the core structure of anthraquinone. We identified 11 crucial gene families in the shikimate pathway (Supplementary Table S17). In addition, compared with C. canephora, the key gene DHQS in the shikimate pathway was expanded to enhance the ability to produce 3-dehydroquinate (DHQ). On the other hand, the chlP gene number was also expanded, which may contribute to terpenoid-quinone biosynthesis. We also identified 14 important gene families in the MVA and MEP pathways (Fig. 5a and Supplementary Table S17). Almost every node in the MEP pathway had only one gene copy, while the crucial genes ACAT, HMGR and PMK in the MVA pathway had two or three gene copies. Gene expression analysis showed that most MEP pathway genes had high expression in leaves, while genes in the MVA pathway were expressed in various tissues.

a Anthraquinone and terpenoid biosynthesis pathways. b Polysaccharide biosynthesis pathway. c Part of the starch and sucrose metabolism pathway. Various color blocks represent the normalized gene expression levels of all genes encoding in different tissues. Heat maps of these genes were plotted using the pheatmap package. Red dots represent gene family expansion in M. officinalis
Functional gene evolution contributed to the formation of terpenoids
Terpenoid biosynthesis starts from the terpenoid backbone biosynthesis pathway. Isopentenyl pyrophosphate (IPP) and DMAPP are common precursors for terpenoid biosynthesis in plants and are mainly formed by the MVA and MEP pathways. Geranylgeranyl diphosphate (GGPP) is the direct precursor substrate of diterpene biosynthesis, which GGPPS catalyzes. In the M. officinalis genome, we found that GGPPS has ten gene copies, and tandem duplication was found on Chr6 (Fig. 6 and Supplementary Table S17). Gene expression analysis showed that GGPPS might have potential functional divergence; for example, evm.model.LG06.1002 was relatively highly expressed in AR, but evm.model.LG06.1003 showed no expression in any tissues (Supplementary Table S17).

Gene families related to anthraquinone and terpenoid biosynthesis pathways (chlP, CYP82G1, DHQS, GGPPS, TPS04 and TPS-Cin) and sugar metabolism (sacA, bglB, AMY, BGL, GN5/6, UGDH, SPS, TPS and TREH)
Terpene synthase (TPS) is vital for terpenoid synthesis and uses geranyl diphosphate (GPP), GGPP, and farnesyl diphosphate (FPP) as direct precursors to synthesize monoterpenes, diterpenes, sesquiterpenes, and triterpenes. TPS proteins were identified by using the Pfam domain models PF03936 and PF01397 with an E-value cutoff of 1e−5. We identified 41 TPS family genes in the M. officinalis genome. Based on the phylogenetic tree generated by aligning the TPS proteins of M. officinalis, C. canephora, and A. thaliana, we divided the M. officinalis TPS genes into seven groups (Supplementary Fig. S7a and Supplementary Table S18). The TPS-a (14 genes) and TPS-b (14 genes) groups contained more genes, while the TPS-g group comprised only two genes. We also found that the TPS-a group had tandem duplication on Chr2 (10 genes), and the TPS-b group had tandem duplication on Chr2 (6 genes) and Chr6 (6 genes) (Supplementary Table S18). Thus, tandem duplication is responsible for TPS-Cin and TPS04 gene family expansion in M. officinalis after its split from C. canephora (Fig. 6). In addition, we noticed that the expression patterns of most genes in the same group were similar. Nevertheless, TPS-a was mainly expressed in AR, and TPS-b showed high expression in stalks, which may be related to the tissue-specific localization of substance synthesis (Supplementary Fig. S7b and Supplementary Table S18).
Iridoids are the major terpenoids in M. officinalis and are monoterpene analogs. In the iridoid biosynthesis pathway, one GES (TPS-g subfamily), fifteen G10Hs, six 10HGOs, three ISs, four 7-DLSs and twelve 7-DLGTs were identified (Fig. 5a and Supplementary Table S17). Based on the chromosome location, we found that these functional genes (G10H, 10HGO, IS, 7-DLS, and 7-DLGT) may have undergone tandem duplication in the M. officinalis genome. Interestingly, this duplication also existed in C. canephora, which suggested that the duplication of these gene families may have occurred before the speciation of M. officinalis and C. canephora.
Significant expansion of genes related to sugar metabolism
As shown in Fig. 5b, we deduced the synthesis pathway of polysaccharides in M. officinalis based on the enzymes involved in carbon metabolism in the pathways “starch and sucrose metabolism” and “amino and nucleotide sugar metabolism”. In M. officinalis, nine genes encoding sacA, four genes encoding malZ, six genes encoding scrK, and 12 genes encoding HK were identified, most of which were highly expressed in leaves and stalks (Fig. 5b and Supplementary Table S19). We also identified 44 genes encoding nucleotide-diphospho-sugar interconversion enzymes, most of which showed diverse expression patterns in different tissues, indicating the complexity of the regulation of polysaccharide biosynthesis (Fig. 5b and Supplementary Table S19). The sacA gene catalyzes the formation of D-fructose and Glc-6P as precursors. Two tandem repeat blocks were found on Chr1 and Chr3 (Fig. 6). UGDH is responsible for the biosynthesis of UDP-GlcA, which might be the restrictive precursor of other nucleotide-diphospho-sugars, such as UDP-Gal, UDP-d-Xyl and UDP-l-Ara. We found that the UGDH gene in M. officinalis was expanded compared to that in C. canephora, which might have contributed to the formation of UDP-GlcA (Fig. 6 and Supplementary Table S20).
We also paid particular attention to other expanded gene families related to the starch and sucrose metabolism pathways (Fig. 5c). Based on their function, these extended genes catalyze the formation of glucose from other sugars, which is one of the important substrates for glycolysis and polysaccharide synthesis. A significant tandem duplication event of BGL genes was identified in M. officinalis on Chr1, Chr2, Chr4, contig9, and contig 17 (Fig. 6). For the bglB genes, similar tandem repeats were found on Chr3, Chr9, and Chr11 (Fig. 6 and Supplementary Table S20). The AMY genes can decompose starch into maltose and then form glucose under the catalysis of malQ and malZ. Tandem duplication of AMY genes was also found on Chr6 (Fig. 6 and Supplementary Table S20). These results suggested that tandem duplication is responsible for the expansion of these genes after the separation between M. officinalis and C. canephora.
Discussion
M. officinalis is one of the top four most famous southern herbs in China. Nevertheless, in the past, research on M. officinalis has mainly focused on its pharmacology. Because of the impediments associated with the sexual reproduction of this species and the exhaustion of its wild resources, germplasm resources are very limited15. Additionally, the lack of genomic information seriously hinders genetic research on M. officinalis. Therefore, obtaining genomic information on M. officinalis will enable potential genetic improvements and the development of molecular breeding resources. However, a genome survey showed that its genome was highly heterozygous (Supplementary Fig. S1). High repetition rates and high heterozygosity are challenges for high-quality genome assembly23. To overcome this issue, we combined third-generation sequencing and Hi-C technology to assemble the M. officinalis genome. Here, we present a high-quality chromosome-scale genome sequence for M. officinalis, with a scaffold N50 of 40.97 Mb, which is higher than that of other medicinal plants, such as S. baicalensis (408.14 Mb, N50 33.2 Mb)19. A total of 97.02% complete BUSCO core genes were detected in our assembly, which suggested that the quality of this reference genome is comparable to that of the published highly heterozygous T. wilfordii genome (95.10%)24. In brief, the assembly of M. officinalis is relatively accurate and complete, which will provide a valuable genome resource for understanding the evolution, active ingredient biosynthesis, and genetic improvement of this species.
The genus Morinda (Rubiaceae), which includes 102 species, is distributed in tropical, subtropical, and temperate regions of the world2. However, there are few reports on genome research of the species in this genus. We found that 82.77% of annotated genes were matched to the proteins of the genus Coffea (Rubiaceae). This indicates the lack of reference genome sequences and limited public data on the genus Morinda. As one of the members of this genus, the genome information of M. officinalis can fill this gap and promote evolutionary research. Ks analysis found that M. officinalis shared only an ancient WGD event with C. canephora and V. vinifera, and no recent WGD event occurred (Fig. 3c). The M. officinalis chromosomal regions showed a one-to-one correspondence with V. vinifera; similar results were also found between C. canephora and V. vinifera25. WGD events can cause plant genome size variation, chromosomal rearrangement, gene family expansion and species evolution26,27. We found a high collinearity relationship between M. officinalis and C. canephora chromosomes, which may be because they have not undergone recent WGD events or large-scale chromosomal variation after species divergence (Supplementary Fig. S6).
M. officinalis diseases are one of the critical factors that affect yield and quality. Stem rot is a common destructive disease of M. officinalis, often occurring at the stem base17. We identified 59 expanded and 25 contracted genes associated with plant–pathogen interactions in the M. officinalis genome (Supplementary Table S14 and Supplementary Fig. S8). These extended genes are located on all chromosomes of M. officinalis, and the KCS gene (K15397) and CML gene (K13448) have undergone tandem duplication on Chr5 and Chr6, respectively (Supplementary Fig. S8). This may be a unique adaptative mechanism evolved by M. officinalis in response to changes in various pathogens during natural selection. Based on the gene expression patterns, we found increased expression of 2,741 DEGs in the stalks, and functional enrichment showed that these genes were significantly associated with defense responses, such as “MAPK signaling pathway-plant” and “plant–pathogen interaction” (Fig. 4b and Supplementary Table S16). These genes (e.g., FLS2, MPK3/6, and WRKY22) are involved in signal perception, cascade amplification, transmission, and regulation of downstream functional gene expression, which plays a crucial role in plant disease resistance by regulating multiple defenses28,29,30. Therefore, our results provide insights into the molecular mechanism of the interaction between M. officinalis and pathogens.
As medicinal and edible plants, M. officinalis contains medicinally active compounds, mainly including anthraquinones, iridoids, and polysaccharides1,2,10. Although genes involved in anthraquinone, iridoid, and polysaccharide biosynthesis have been identified in other medicinal plants, such as Ophiorrhiza pumila, Senna tora, Gardenia jasminoides, and Artemisia sphaerocephala, the biosynthesis pathways in M. officinalis are still unclear31,32,33,34. Previous studies have shown that there are two metabolic pathways in plant anthraquinone biosynthesis. The shikimate/o-succinylbenzoic acid pathway mainly exists in Rubiaceae plants, while the polyketide pathway mainly exists in fungi and other plants, such as Leguminosae, Rhamnaceae, and Polygonaceae35,36. Terpenoids are the main active ingredients of many medicinal plants, such as Andrographis paniculata, Gynostemma pentaphyllum and Gardenia jasminoides37,38,39. The synthesis of terpenoids starts from the common precursors IPP and DMAPP, and then TPS converts the corresponding substrates to form structurally diverse monoterpenes, diterpenes, sesquiterpenes, etc.40. Polysaccharides are important bioactive components with various activities. Previous studies have shown that the polysaccharides in M. officinalis are mainly composed of glucose and fructose10,11. In this work, we identified candidate genes related to the biosynthesis of these active components in M. officinalis and analyzed their expression patterns in different tissues (Fig. 5a, b). We also found that some essential genes involved in the synthesis of these active ingredients, such as DHQS, GGPPS, TPS-Clin, TPS04, sacA, and UGDH, expanded in M. officinalis (Fig. 6). The expansion of key genes in metabolic pathways is beneficial to the synthesis and accumulation of active components and is a common event in medicinal plants during the evolutionary process41. Notably, we found that the expression patterns of some expanded genes with the same function were diverse, indicating a potential functional divergence of these gene families.
The roots of M. officinalis serve as an effective agent in traditional Chinese medicine, and there are two main stages in plant growth and development in this species: the growth of aboveground tissues and the growth of roots. Therefore, the vines need to be manually cut to promote the expansion of the roots after three years of cultivation, but this process may affect photosynthesis. Glucose is an important substrate for glycolysis and the TCA cycle, which provides energy for biological activities and intermediates for other metabolic processes42. Interestingly, we found that gene families involved in converting other sugars to glucose, such as BGL, AMY, and TREH, expanded significantly (Fig. 6). We speculate that these evolutionary genes in M. officinalis may need to compensate for glucose deficiency. Although we have identified related candidate genes, their functions and evolutionary mechanisms should be explored in future work.
Conclusions
In this work, we first report the high-quality chromosome-scale reference genome of M. officinalis. Genome evolution showed that M. officinalis shared an ancient WGD event with C. canephora and V. vinifera. We further used high-quality genome information to identify candidate genes for terpenoid, anthraquinone, and polysaccharide biosynthesis. We found that the functional genes related to pathogen resistance and active component biosynthesis were expanded in the M. officinalis genome. Overall, this high-quality reference genome provides insights into genome evolution and active component biosynthesis in M. officinalis. Our research also lays the foundation for further studies for genetic improvement and breeding, not only in M. officinalis but also in other Morinda species.
Materials and methods
Plant materials and sequencing
Root, leaf, and stem samples of “Gaoji 3”, a cultivated variety of M. officinalis, were collected from the Gaoyao District (Zhaoqing city, 28°28′–28°66′ N, 85°13′–85°28′ E) in Guangdong Province, China. Because of its unique geography and environment, the Gaoyao District of Zhaoqing City is considered the authentic production area of M. officinalis. “Gaoji 3”, with many excellent cultivation characteristics, including high yield, high quality, and disease resistance, is the main variety of M. officinalis grown in Gaoyao District.
High-quality genomic DNA was extracted using a QIAGEN® Genomic Kit (QIAGEN, Germany). The integrity of the DNA was checked by 0.75% agarose gel electrophoresis. The purity and concentration of the DNA were analyzed by using a NanoDrop 2000 spectrophotometer (ThermoFisher Scientific, USA) and Qubit Fluorometer (ThermoFisher Scientific, USA). A paired-end library was constructed and sequenced using the MGISEQ-2000 (BGI, Shenzhen, China) and Illumina NovaSeq 6000 platforms (Illumina, San Diego, USA) to generate short sequencing reads. We constructed DNA libraries for long-read single-molecule sequencing and sequenced them on the Nanopore PromethION platform. Total RNA was extracted from the roots, stems, and leaves using an RNAprep Pure Plant Kit (TIANGEN Biotech, China). The RNA libraries were prepared using a TruSeq RNA Library Kit (Illumina, CA, USA) and then sequenced on the Illumina NovaSeq 6000 platform.
The quality of the short sequencing reads was estimated using the FastQC tool, and the adapter sequences, contamination, PCR duplicates, and low-quality reads (reads with more than 30 low-quality bases or 5% unknown bases) were removed using fastp43,44. For the Nanopore data, long raw reads were converted into fastq format after base calling by using the Guppy tool45. Reads with a mean_qscore_template value greater than seven were retained46. NextDenovo (https://github.com/Nextomics/NextDenovo), with the parameters read_cutoff = 1 kb and seed-cutoff = 37 kb, was employed to ensure further correction and assembly. These clean data were used for further assembly and subsequent analysis.
Genome survey
To estimate the M. officinalis genome characteristics, we used a K-mer-based method to estimate the genome size and heterozygosity. Approximately 61.4 Gb (~127×) clean short reads were generated and used for 17 K-mer analyses (Supplementary Table 1). The frequency distribution of 17 K-mers was counted by Jellyfish software47. Then, the K-mer depth distribution curve was calculated to estimate the genome size. To further estimate the heterozygosity rate, the genome of Arabidopsis thaliana was used to simulate the corresponding depth of short-read data, and K-mer curve fitting was carried out under different gradient heterozygosity rates. The heterozygosity rate was estimated according to K-mer curve fitting.
Genome and chromosome assembly
After quality control, a total of 62.92 Gb (~130×) corrected Nanopore long reads were generated with an average length of 22.39 Kb and N50 of 30.16 Kb. These qualified long reads were assembled by using NextGraph with default parameters to obtain the preliminary assembled genome sequence. The Nanopore long reads were mapped to the preliminary assembled genome using minimap2 with the parameters “-x map-ont” and subjected to three rounds of polishing using Nextpolish with default parameters48,49. Furthermore, to improve accuracy, the clean paired-end reads were mapped to the genome sequence using bwa to polish the assembled contigs with Pilon (iterative correction of four times)50,51. Finally, Redundant software was employed to resolve redundancy in the assembly with the parameters “–identity 0.8–overlap 0.8” to obtain the final nonredundant genome52.
To further anchor the contigs to chromosomes, we used fresh young leaves to construct a Hi-C library using a NEBNext Ultra II DNA Library Prep Kit. Approximately 55.8 Gb of clean data were generated for Hi-C analyses by the Illumina NovaSeq 6000 platform. These clean reads were mapped to the M. officinalis genome using Bowtie2 software (v2.3.2) with the parameters “–very-sensitive -L 30” to obtain the uniquely mapped paired-end reads53. After filtering out the invalid interaction pairs, including dangling end paired-end reads, self-circle paired-end reads, and dumped paired-end reads, valid paired-end reads were identified and separated by using HiC-Pro (v2.7.8)54. We then used LACHESIS software to cluster and reorder the contigs into pseudochromosomes with the following parameters: cluster min re sites = 100, cluster max link density = 2.5, cluster noninformative ratio = 1.4, order min n res in trunk = 60, and order min n res in shreds = 6055. Finally, the order and direction of the contigs on the pseudochromosomes were evaluated and adjusted by examining their interactions in the Hi-C heatmap.
Evaluation of the assembled genome
We used multiple methods to assess the accuracy and completeness of the assembled genome. First, the paired-end reads were mapped to the genome to evaluate its completeness using bwa with the default parameters. RNA-seq data from different tissues (leaf, stalk, and root) were also aligned to the reference genome to obtain the mapping rate using HISAT2 with the default settings56. Second, GC depth scatter plots were used to evaluate any contamination in the sequencing data. Finally, the accuracy and completeness of the genome assembly were evaluated by using BUSCOs to identify the single-copy genes in the assembled genome with the Embryophyta_odb10 database57.
Repeat element identification
The repetitive sequences in the genome can be divided into two main categories: tandem repeats and transposable elements. We used two software programs, GMATA and Tandem Repeats Finder, to search for tandem repeats in the whole genome with default parameters58,59. Homology alignment and de novo searches were combined to identify transposable elements. RepeatModeler was used for de novo searching for repetitive sequences, which were then classified with Teclass60,61. We identified the repeats through a homology-based repeat search using Repbase62. We also used MITE-hunter to discover the small transposon called MITE63. LTR_finder and LTR_harvest software were employed to identify the LTRs, and LTR_retriever was used to integrate these results to obtain an LTR retrotransposon library of M. officinalis64,65,66.
Noncoding RNA prediction
The tRNA genes were predicted using tRNAscan-SE, and other noncoding RNAs, including rRNA, snRNA, and miRNA, were predicted by comparison with the Rfam database using Infernal software with the default parameters67,68,69. At the same time, we also used RNAmmer software to construct models to predict rRNA and its various subunits70.
Gene prediction and functional annotation
Transcriptome-based, homology-based, and ab initio prediction methods were combined to predict gene models in the M. officinalis genome. To improve gene prediction, RNA libraries were prepared from mixed fresh leaf, stem, and root tissues, and finally 33.43 Gb clean data were generated. For homology-based annotation, the protein sequences of C. canephora25, C. Arabica71, C. roseus72, and A. thaliana73 were downloaded and aligned against the M. officinalis genome using GeMoMa74. For transcriptome-based prediction, the non-redundant transcripts were aligned to the reference genome to obtain gene structures using PASA75. Then, TransDecoder was used to search the longest open reading frames according to the PASA results76. We chose 3000 genes with the highest alignment scores (identity >95%) as the training sets for the AUGUSTUS model to generate a generalized hidden Markov probability model for ab initio gene prediction77. Finally, we integrated the gene models from the three approaches with EvidenceModeler, and TransposonPSI was used to remove genes containing transposable elements to generate the final consensus gene models78,79.
Functional annotation of the protein-coding genes was carried out by using Blastp with a cutoff E-value of 1e−5 with different public databases. The functions of the genes were predicted and classified using the KOG, NR, and UniProtKB/SwissProt databases. The GO database classified and annotated genes according to three categories: biological processes, cellular components, and molecular functions. We used InterProScan software to identify protein domains by matching them against Pfam database entries to obtain GO terms80. Pathway annotation was performed with the KEGG database with an E-value ≤ 1e−5.
Gene families and phylogenetic analysis
We collected the protein sequences of M. officinalis and nine other species, including A. thaliana, Capsicum chinense, C. roseus, C. canephora, Erythranthe guttata, Olea europaea, Solanum lycopersicum, Striga asiatica, and V. vinifera, for evolutionary analysis. An all-to-all BLASTP analysis of the protein sequences of all species was performed with an E-value ≤ 1e−5. Subsequently, orthologous genes, paralogous genes, and single-copy homologous genes were identified using OrthoMCL software with the default parameters81.
Phylogenetic construction was performed based on the single-copy orthologous genes from the ten species. We used MAFFT software to align the protein sequences and extracted the conserved sites from the alignments using Gblocks82,83. We used V. vinifera and A. thaliana as an outgroup and performed 1000 bootstrap replicates to construct a phylogenetic tree by RaxML84. The divergence time of each species was estimated based on the Bayesian relaxed molecular clock approach using MCMCTREE of the PAML package85. According to the results of the gene families and phylogenetic tree, λ values were estimated with CAFE software to predict the expansion or contraction of gene families of different species in each evolutionary branch based on a stochastic birth and death process model86. Gene families with a p-value smaller than 0.05 were considered significantly expanded or contracted.
Positive selection genes and WGD analysis
Based on the results of single-copy genes, Codeml software was used to calculate the selection pressure to identify the positively selected genes of M. officinalis (p-value ≤ 0.05)87. We performed multiple sequence alignment to extract the conserved paralogs of the protein sequences of M. officinalis by using BLASTP (E-value ≤ 1e−5). MCScan was used to identify collinearity blocks, and Ks values were calculated by PAML to predict WGD events88.
Transcriptome profiling analysis
A total of 108.94 Gb clean reads were produced from the leaves, stalks, and roots at different developmental stages of M. officinalis. These reads were aligned to the reference genome by using HISAT256. The fragments per kilobase of transcript per million fragments mapped (FPKM) were calculated to estimate the expression level of genes. EdgeR was used to analyze the significantly DEGs with an FDR ≤ 0.05 and an absolute log2 (fold change) ≥1 as the threshold89.
Data availability
The sequencing data, including the MGI genome data (SRA accession: SAMN14599083) and Hi-C data (SRA accession: SAMN14599083), are available in the NCBI Sequence Read Archive (SRA) database under BioProject accession number PRJNA625354.
References
Lim, T. K. Edible Medicinal and Non-Medicinal Plants Vol. 5 (Springer, 2013).
Zhang, J. H. et al. Morinda officinalis how—a comprehensive review of traditional uses, phytochemistry and pharmacology. J. Ethnopharmacol. 213, 230–255 (2018).
Wu, Z. Q. et al. Effect of bajijiasu isolated from Morinda officinalis FC how on sexual function in male mice and its antioxidant protection of human sperm. J. Ethnopharmacol. 164, 283–292 (2015).
Bao, L. et al. Anthraquinone compounds from Morinda officinalis inhibit osteoclastic bone resorption in vitro. Chem. Biol. Interact. 194, 97–105 (2011).
Zhao, X., Wei, J. & Yang, M. Simultaneous analysis of iridoid glycosides and anthraquinones in Morinda officinalis using UPLC-QqQ-MS/MS and UPLC-Q/TOF-MSE. Molecules 23, 1070 (2018).
Yong, J., Lu, C., Huang, S. & Wu, X. Chemical components isolated from the roots of Morinda officinalis. Chem. Nat. Compd. 51, 548–549 (2015).
Malik, E. M. & Müller, C. E. Anthraquinones as pharmacological tools and drugs. Med. Res. Rev. 36, 705–748 (2016).
PHAKHODEE, W. Distribution of naturally occurring anthraquinones, iridoids and flavonoids from Morinda genus: chemistry and biological activity. Walailak J. Sci. Technol. 9, 173–188 (2012).
Choi, J. et al. Antinociceptive anti-inflammatory effect of monotropein isolated from the root of Morinda officinalis. Biol. Pharm. Bull. 28, 1915–1918 (2005).
Zhang, H. L. et al. Structural characterization and anti-fatigue activity of polysaccharides from the roots of Morinda officinalis. Int. J. Biol. Macromol. 44, 257–261 (2009).
Zhu, M. et al. Extraction of polysaccharides from Morinda officinalis by response surface methodology and effect of the polysaccharides on bone-related genes. Carbohydr. Polym. 85, 23–28 (2011).
Li, Y. F., Gong, Z. H., Yang, M., Zhao, Y. M. & Luo, Z. P. Inhibition of the oligosaccharides extracted from Morinda officinalis, a Chinese traditional herbal medicine, on the corticosterone induced apoptosis in PC12 cells. Life Sci. 72, 933–942 (2003).
Yan, C. et al. Identification and characterization of a polysaccharide from the roots of Morinda officinalis, as an inducer of bone formation by up-regulation of target gene expression. Int. J. Biol. Macromol. 133, 446–456 (2019).
Li, X. et al. Research of Morinda officinalis how’s oligosaccharide extraction and antidepressant effects. Bulgar. Chem. Commun. 49, 162–167 (2017).
Zhang, R. et al. Investigation on germplasm resources of Morinda officinalis how. Mod. Chin. Med 18, 482–487 (2016).
Liu, J., Ding, P., Zhan, R. T. & Chen, W. W. Resource survey of medicinal plant of Morinda officinalis how in Guangdong and Fujian Provinces. J. Guangzhou Univ. Trad. Chin. Med. 26, 485–487 (2009).
Luo, M., Shu, Y., Zhang, W. & Dong, Z. Research progress of Morinda officinalis disease. J. Agric. Sci. 8, 1312–1317 (2018).
Yang, J., Jia, M. & Guo, J. Functional genome of medicinal plants. Mol. Pharmacogn. 11, 191–234 (2019).
Zhao, Q. et al. The reference genome sequence of Scutellaria baicalensis provides insights into the evolution of wogonin biosynthesis. Mol. Plant 12, 935–950 (2019).
Kang, M. et al. A chromosome-scale genome assembly of Isatis indigotica, an important medicinal plant used in traditional Chinese medicine. Hortic. Res. 7, 1–10 (2020).
Li, L. F., Cushman, S. A., He, Y. X. & Li, Y. Genome sequencing and population genomics modeling provide insights into the local adaptation of weeping forsythia. Hortic. Res. 7, 1–12 (2020).
Liu, X. et al. The genome of medicinal plant Macleaya cordata provides new insights into benzylisoquinoline alkaloids metabolism. Mol. Plant 10, 975–989 (2017).
Gao, S. et al. A high-quality reference genome of wild Cannabis sativa. Hortic. Res. 7, 1–11 (2020).
Tu, L. et al. Genome of Tripterygium wilfordii and identification of cytochrome P450 involved in triptolide biosynthesis. Nat. Commun. 11, 1–12 (2020).
Denoeud, F. et al. The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science 345, 1181–1184 (2014).
Christianson, D. W. Roots of biosynthetic diversity. Science 316, 60–61 (2007).
El Baidouri, M. & Panaud, O. Comparative genomic paleontology across plant kingdom reveals the dynamics of TE-driven genome evolution. Genome Biol. Evol. 5, 954–965 (2013).
Zou, Y. et al. Transcriptional regulation of the immune receptor FLS2 controls the ontogeny of plant innate immunity. Plant Cell 30, 2779–2794 (2018).
Meng, X. et al. Phosphorylation of an ERF transcription factor by Arabidopsis MPK3/MPK6 regulates plant defense gene induction and fungal resistance. Plant Cell 25, 1126–1142 (2013).
Saand, M. A., Xu, Y. P., Li, W., Wang, J. & Cai, X. Z. Cyclic nucleotide gated channel gene family in tomato: genome-wide identification and functional analyses in disease resistance. Front. Plant Sci. 6, 303 (2015).
Yamazaki, M. et al. Coupling deep transcriptome analysis with untargeted metabolic profiling in Ophiorrhiza pumila to further the understanding of the biosynthesis of the anti-cancer alkaloid camptothecin and anthraquinones. Plant Cell Physiol. 54, 686–696 (2013).
Kang, S. H. et al. Genome-enabled discovery of anthraquinone biosynthesis in Senna tora. Nat. Commun. 11, 1–11 (2020).
Ye, P. et al. Transcriptome analysis and targeted metabolic profiling for pathway elucidation and identification of a geraniol synthase involved in iridoid biosynthesis from Gardenia jasminoides. Ind. Crops Prod. 132, 48–58 (2019).
Han, X. et al. Transcriptome analysis reveals the molecular mechanisms of mucilage biosynthesis during Artemisia sphaerocephala seed development. Ind. Crops Prod. 145, 111991 (2020).
Han, Y. S., Van der Heijden, R. & Verpoorte, R. Biosynthesis of anthraquinones in cell cultures of the Rubiaceae. Plant Cell Tissue Organ Cult. 67, 201–220 (2001).
Liang, W. et al. Research progress on synthesis of anthraquinones based on shikimic acid/o-succinylbenzoic acid pathway. Chin. Trad. Herb. Drugs 51, 1939–1950 (2020).
Sun, W. et al. The genome of the medicinal plant Andrographis paniculata provides insight into the biosynthesis of the bioactive diterpenoid neoandrographolide. Plant J. 97, 841–857 (2019).
Xu, S., Yao, S., Huang, R., Tan, Y. & Huang, D. Transcriptome-wide analysis of the AP2/ERF transcription factor gene family involved in the regulation of gypenoside biosynthesis in Gynostemma pentaphyllum. Plant Physiol. Biochem. 154, 238–247 (2020).
Li, H. B. et al. Two new iridoid glycosides from the fruit of Gardenia jasminoides. Nat. Prod. Res. 2015, 1–7 (2020).
Xia, Z. et al. Chromosome-scale genome assembly provides insights into the evolution and flavor synthesis of passion fruit (Passiflora edulis Sims). Hortic. Res. 8, 1–14 (2021).
Xu, Z. et al. Comparative genome analysis of Scutellaria baicalensis and Scutellaria barbata reveals the evolution of active flavonoid biosynthesis. Genomics Proteomics Bioinformatics 18, 230–240 (2020).
Fernie, A. R., Carrari, F. & Sweetlove, L. J. Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. plant Biol. 7, 254–261 (2004).
Patel, R. K. & Jain, M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE 7, e30619 (2012).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Wick, R. R., Judd, L. M. & Holt, K. E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 20, 129 (2019).
Senol Cali, D., Kim, J. S., Ghose, S., Alkan, C. & Mutlu, O. Nanopore sequencing technology and tools for genome assembly: computational analysis of the current state, bottlenecks and future directions. Brief Bioinformatics 20, 1542–1559 (2019).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Li, H. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110 (2016).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long read assembly. Bioinformatics 36, 2253–2255 (2020).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997 (2013).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Pryszcz, L. P. & Gabaldón, T. Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 44, e113–e113 (2016).
Langmead, B. & Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125 (2013).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Wang, X. & Wang, L. GMATA: an integrated software package for genome-scale SSR mining, marker development and viewing. Front. Plant Sci. 7, 1350 (2016).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Bedell, J. A., Korf, I. & Gish, W. MaskerAid: a performance enhancement to RepeatMasker. Bioinformatics 16, 1040–1041 (2000).
Abrusán, G., Grundmann, N., DeMester, L. & Makalowski, W. TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25, 1329–1330 (2009).
Jurka, J. et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Han, Y. & Wessler, S. R. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 38, e199 (2010).
Xu, Z. & Wang, H. LTR-FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18 (2008).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Lowe, T. M. & Chan, P. P. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57 (2016).
Gardner, P. P. et al. Rfam: updates to the RNA families database. Nucleic Acids Res. 37, D136–D140 (2009).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
De Nardi, B. et al. Differential responses of Coffea arabica L. leaves and roots to chemically induced systemic acquired resistance. Genome 49, 1594–1605 (2006).
Franke, J. et al. Gene discovery in Gelsemium highlights conserved gene clusters in monoterpene indole alkaloid biosynthesis. ChemBioChem 20, 83–87 (2019).
Kaul, S. et al. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815 (2000).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89–e89 (2016).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Urasaki, N. et al. Draft genome sequence of bitter gourd (Momordica charantia), a vegetable and medicinal plant in tropical and subtropical regions. DNA Res. 24, 51–58 (2017).
Hunter, S. et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215 (2009).
Li, L., Stoeckert, C. J. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Yang, Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13, 555–556 (1997).
De Bie, T., Cristianini, N., Demuth, J. P. & Hahn, M. W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006).
Gao, F. et al. EasyCodeML: a visual tool for analysis of selection using CodeML. Ecol. Evol. 9, 3891–3898 (2019).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49–e49 (2012).
Robinson, M. D., McCarthy, D. J. & Smyth, G K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Acknowledgements
This project was supported by a study on the cultivation of the six new varieties of Lingnan Traditional Chinese Medicine (2020B020221001), Scientific Innovation Strategy Construction of the High-level Academy of Agriculture Science (R2019PY-JX003), and Southern Medicinal Plants Modern Agricultural Industrial Park of Gaoyao (2018).
Author information
Authors and Affiliations
Contributions
S.Y. and M.Z. designed and supervised the project. S.C., Y.M., Y.G., Z.L., and M.S. prepared the samples. Y.X., S.X., and J.W. analyzed the data. J.W. and S.X. wrote the manuscript, and S.Y. and M.Z. revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, J., Xu, S., Mei, Y. et al. A high-quality genome assembly of Morinda officinalis, a famous native southern herb in the Lingnan region of southern China. Hortic Res 8, 135 (2021). https://doi.org/10.1038/s41438-021-00551-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-021-00551-w
This article is cited by
-
Applications of some advanced sequencing, analytical, and computational approaches in medicinal plant research: a review
Molecular Biology Reports (2024)
-
Genomic-wide identification and expression analysis of R2R3-MYB transcription factors related to flavonol biosynthesis in Morinda officinalis
BMC Plant Biology (2023)
-
Insights into glucosinolate accumulation and metabolic pathways in Isatis indigotica Fort.
BMC Plant Biology (2022)
