
Abstract
The genus Aquilegia (Ranunculaceae) has been cultivated as ornamental and medicinal plants for centuries. With petal spurs of strikingly diverse size and shape, Aquilegia has also been recognized as an excellent system for evolutionary studies. Pollinator‐mediated selection for longer spurs is believed to have shaped the evolution of this genus, especially the North American taxa. Recently, however, an opposite evolutionary trend was reported in an Asian lineage, where multiple origins of mini- or even nonspurred morphs have occurred. Interesting as it is, the lack of genomic resources has limited our ability to decipher the molecular and evolutionary mechanisms underlying spur reduction in this special lineage. Using long-read sequencing (PacBio Sequel), in combination with optical maps (BioNano DLS) and Hi–C, we assembled a high-quality reference genome of A. oxysepala var. kansuensis, a sister species to the nonspurred taxon. The final assembly is approximately 293.2 Mb, 94.6% (277.4 Mb) of which has been anchored to 7 pseudochromosomes. A total of 25,571 protein-coding genes were predicted, with 97.2% being functionally annotated. When comparing this genome with that of A. coerulea, we detected a large rearrangement between Chr1 and Chr4, which might have caused the Chr4 of A. oxysepala var. kansuensis to partly deviate from the “decaying” path that was taken before the split of Aquilegia and Semiaquilegia. This high-quality reference genome is fundamental to further investigations on the development and evolution of petal spurs and provides a strong foundation for the breeding of new horticultural Aquilegia cultivars.
Similar content being viewed by others


Introduction
The genus Aquilegia (Ranunculaceae), commonly known as columbine, consists of ~70 species that are widely distributed in the temperate zones of the Northern Hemisphere1. Numerous species and varieties of this genus have been cultivated as garden ornamentals for centuries due to their attractive flowers with unusual characteristics, including petaloid sepals and petal spurs of diverse shape and size2,3,4. Particularly, the length of spurs (∼1–16 cm) varies dramatically, matching the tongue length of the corresponding pollinators; this has made the genus a model system for research on pollinator-driven diversification2,5. In fact, the multiple origins of species with longer petal spurs from those with shorter ones in the North American Aquilegia clade have become textbook examples of pollinator shift-mediated adaptive evolution5,6,7. Recently, however, one Asian Aquilegia lineage was revealed to have experienced multiple origins of mini- or even nonspurred morphs, indicating an opposite trend of spur evolution8,9. The genus, therefore, is also an excellent system for the study of spur reduction. Deciphering the developmental and evolutionary mechanisms underlying these morphological changes would not only facilitate our understanding of species diversification but also provide a good foundation for the breeding of promising horticultural Aquilegia cultivars.
Genomic resources are essential and attainable for developmental and evolutionary studies. To date, only one genome in the Aquilegia genus has been sequenced (A. coerulea reference genome v3.1)10. A. coerulea has flowers with long petal spurs and belongs to the North American clade10,11; its genomic resources have facilitated numerous studies concerning spur elongation, adaptation, and speciation12,13,14,15,16,17,18. However, the application of these resources to address the problems of spur reduction is limited because A. coerulea diverged from the Asian lineage approximately 4.8 million years ago (Mya)11. Moreover, several lines of evidence suggest a “decaying” nature of Aquilegia Chr4, which has likely been evolving under reduced purifying and/or background selection and has a strikingly higher level of polymorphism than the rest of the genome10. This further aggravates the difficulty in referring to the genome of a phylogenetically far-related species.
Here, we report the genome of A. oxysepala var. kansuensis (Fig. 1), a species that is sister to the nonspurred taxon in Asia8. We assembled a chromosome-scale reference genome by long-read sequencing (PacBio Sequel system), optical map (BioNano DLS) scaffolding, and further anchoring the scaffolds to pseudochromosomes using Hi–C. Comparison of the A. coerulea and A. oxysepala var. kansuensis genomes revealed a large chromosomal rearrangement between Chr1 and Chr4, and we cataloged the species-specific genes. Notably, we found that, unlike Chr4 in other Aquilegia taxa, this chromosome in A. oxysepala var. kansuensis is not evolving entirely under reduced purifying selection due to the rearrangement event.

Images of A. oxysepala var. kansuensis
Results
Genome size and heterozygosity estimation
We used a single individual of A. oxysepala var. kansuensis that was collected from Yuzhong County, Gansu Province, China, for whole-genome sequencing. We noticed that a new name, A. yangii, has recently been given to plants of this and many other populations19. However, we decided to stay with the original name because the new one is still poorly known. To guide genome sequencing and assembly, we estimated the genome size of A. oxysepala var. kansuensis using flow cytometry20 and K-mer analysis. Briefly, flow cytometry indicated that A. oxysepala var. kansuensis had a genome size of 312 Mb (Fig. S1). For K-mer analysis, we obtained 18.3 Gb short paired-end reads by Illumina sequencing; 18.2 Gb were retained after the removal of low-quality reads (Table S1). Based on the total number of 17-mers and the depth of the main peak in the 17-mer frequency distribution (Fig. S2), we estimated that the genome size and heterozygosity rate of A. oxysepala var. kansuensis were 349 Mb and 0.15%, respectively (Table S2). To ensure we had enough data for genome assembly, we decided to use 349 Mb as a reference for further sequencing.
Sequencing and assembly of the genome
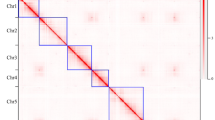
To obtain a high-quality genome, three technologies were applied: PacBio SMRT sequencing (36.7 Gb, ~105×; Table S3, Fig. 2a), BioNano DLS optical mapping (102.1 Gb, ~293×; Table S4, Fig. 2a) and Hi-C mapping (31.9 Gb, ~91×; Table S5, Fig. 2a). The PacBio long-read assemblies showed high contiguity and resulted in a total of only 852 contigs with an N50 of 2.2 Mb, and the longest one was approximately 7.8 Mb (Table S6). After adding the optical mapping data from BioNano DLS, we were able to assemble the contigs into scaffolds. A total of 663 scaffolds (total length = 297.8 Mb) were produced, among which 21 were hybrid scaffolds (total length = 284.8 Mb) encompassing, in most cases, entire chromosome arms. The longest scaffold reached 41.4 Mb, and the N50 was 40.9 Mb. We then anchored all these scaffolds to 7 pseudochromosomes using the Hi–C data in 3D de novo assembly (3D-DNA) software21. Eventually, a high-quality chromosome-level A. oxysepala var. kansuensis assembly (679 scaffolds) was obtained, with 7 pseudochromosomes accounting for 94.6% (277.4/293.2) of the total genome length (Table 1).

a Summary of sequencing data used for genome assembly. Sequencing coverage was calculated based on the genome size obtained from K-mer analysis (349 Mb). For BioNano, effective coverage of the reference is shown. b Hi–C heatmap showing interactions among the 7 pseudochromosomes. c Alignment between the BioNano DLS maps and the final genome assembly
Five approaches were utilized to evaluate the quality of the assembly. First, the Illumina short reads were mapped back to the assembled contigs using Burrows–Wheeler Aligner (BWA) software22. The mapping rate of paired-end reads reached 97.2%, indicating high completeness and accuracy of the final assembly. Second, to assess the completeness of the assembly, we performed benchmarking universal single-copy ortholog (BUSCO)23 analysis by searching against the 1440 conserved single-copy genes in plants and identified 1342 (93.2%) complete BUSCOs (Table 1). Third, when the BioNano assembly consensus genome maps (CMAPs) were aligned to the 7 in silico maps of the A. oxysepala var. kansuensis, a total of 277.3 Mb (unique aligned length) were covered, validating 94.6% of the assembly (Table S7). Fourth, we extracted single nucleotide polymorphisms (SNPs) of the whole-genome using SAMtools24 and found that the proportions of heterozygous and homozygous SNPs were 0.022% and 0.001%, respectively, suggesting high accuracy of the assembly. Finally, we used the LTR assembly index (LAI)—a standard for evaluating the assembly of repeat sequences—to assess assembly continuity25. We found that the LAI score of the A. oxysepala var. kansuensis assembly reached 16.7, which was much higher than that of A. coerulea (LAI = 12.6). Both genomes could be classified as reference quality, similar to the quality of the Arabidopsis TAIR10 genome (LAI = 14.9)25. Taken together, these results suggest that the genome assembly had very high continuity, completeness, and correctness.
Annotation of the genome
Two methods (i.e., homology alignment and de novo annotation) were used to identify repeats in the assembly. Among the five major types of repeats detected (DNA transposon, LINE, LTR, SINE, and unknown), LTRs comprised the largest proportion (35.88%, total length 105.2 Mb). Unknown repeats ranked second, occupying 6.46% (total length 18.9 Mb) of the genome. DNA transposons, LINEs, and SINEs accounted for 2.27%, 1.06%, and 0.01% of the genome, respectively (Fig. 3a, d). Thus, altogether, 45.68% of the genome was predicted to be repeats.

a Distribution of genes and different types of repeat elements: I, Legends of seven pseudochromosomes; II, gene density; III, transposon density; IV, transposable element protein density; and V, tandem repeat density. b Number of genes predicted by different approaches: ab initio (yellow), homology-based (pink) and transcriptome-assisted (gray). c Number of functionally annotated genes based on various databases. d Accumulation history of different kinds of repeat elements
A combination of three methods, including homology-based prediction, ab initio prediction and transcriptome-assisted prediction, was employed to identify protein-coding genes. Sequences from six species were used as references (see Section “Materials and methods”) for homology-based prediction, which generated a total of 22,282 gene models (Fig. 3b). For ab initio prediction, a list of 24,853 putative gene models (Fig. 3b) was obtained by integrating the results from several software packages (see Section “Materials and methods”). To enhance transcriptome-assisted prediction, we performed Iso-Seq and RNA-Seq on various plant tissue samples of different developmental stages (see Section “Materials and methods”). Specifically, Iso-Seq was conducted on five different samples, yielding 46.7 Gb clean data; pooled (equal-amount) total RNA from these samples was also subjected to RNA-Seq, producing 12.8 Gb clean data (Table S8). By applying these data, 18,986 genes were identified (Fig. 3b). The three datasets were then merged together to generate a nonredundant reference gene set containing 25,571 protein-coding genes, with 24,913 on the 7 chromosomes and 658 on the unassigned scaffolds (Fig. 3b). These genes had transcripts that were on average 3,447 bp, including 5 exons with a mean length of 242 bp. We further detected that 24,744, 20,286, 23,692, and 20,374 genes showed significant similarity to known proteins in the NR, Swiss-Prot, InterPro, and KEGG databases, respectively (Fig. 3c). Integration of the four datasets led to the assignment of potential functions to 24,846 (97.2%) of the 25,571 protein-coding genes in the A. oxysepala var. kansuensis genome. We also identified 930 tRNAs, 483 rRNAs (5S, 5.8S, 18S, and 28S), 696 snRNAs, and 275 miRNAs.
Comparison of the genomes of the two Aquilegia taxa
Using MUMmer software (v3.23), we aligned the A. oxysepala var. kansuensis genome assembly with the A. coerulea genome (v3.1)10. For convenience, the chromosomes of these two species will hereafter be referred to as A.ox_chr1–7 and A.co_chr1–7, respectively. The two genomes showed extensive synteny except for one large rearrangement between Chr1 and Chr4 (Fig. 4a, b). Specifically, relative to the A. coerulea genome, there was a reciprocal chromosomal translocation between A.ox_chr1 and A.ox_chr4. To confirm the correct assembly around the breakpoints of the translocated regions, we performed two different analyses. First, we mapped all PacBio long reads to our assembly to determine if there were misjoins caused by a shortage of read mapping evidence near the breaking area. The results showed that the mapping coverage spanning and flanking the area was similar to that in other regions (Fig. S3). Second, we rearranged the two concerned chromosomes, referring to the A. coerulea genome, and thereby created two hypothesized chromosomes (h_A.ox_chr1 and h_A.ox_chr4) that shared chromosome-wide synteny with A.co_chr1 and A.co_chr4, respectively. We then mapped the Hi–C data to h_A.ox_chr1 and h_A.ox_chr4 using Juicer software26. If the two hypothesized chromosomes represented the correct assembly, we would expect a smoother Hi–C interaction heatmap. On the contrary, we observed obvious chromogram discontinuities, indicating misjoins (Fig. S4b). Likewise, when aligning the Hi–C data to the genome of A. coerulea, we also found apparent conflicts on A.co_chr1 and A.co_chr4 (Fig. S4c). These results together suggest that the rearrangement between Chr1 and Chr4 is likely to be real. We further zoomed in the rearrangement region to examine whether there were special genes located around the breakpoints. Adjacent syntenic blocks were compared in detail to determine the exact break sites. As shown in Fig. 4c, the 32.88–33.33 Mb region on A.co_chr1 could be aligned to the 31.47–31.6 Mb of A.ox_chr1, while the downstream 33.31–36.17 Mb aligned to the 29.10–31.00 Mb region of A.ox_chr4. The 29.37–31.18 Mb region of A.co_chr4 was alignable with the 27.08–28.80 Mb region of A.ox_chr4, whereas the downstream 31.77–33.67 Mb shared synteny with the 32.80–33.96 Mb region of A.ox_chr1. From these results, we determined the breakpoint regions, including the 31.60–32.80 Mb region on A.ox_chr1 and the 28.80–29.10 Mb region on A.ox_chr4. These two fragments each contained 16 and 7 genes (Fig. 4c), among which 3 and 2, respectively, had no homologous counterparts in the A. coerulea genome.

a Dot-plot alignment between the two taxa. b Circos plot showing syntenic relationships and chromosomal rearrangements between the two genomes. c Synteny of genes located in and around the breakpoint regions of chromosomes 1 and 4. Red braces stand for the breakpoint regions in A. oxysepala var. kansuensis. d Comparison of gene numbers in different classes of orthologous groups
Compared with the genome of A. coerulea (v3.1), in which 30,023 protein-coding genes were annotated10, the genome of A. oxysepala var. kansuensis contained 4452 fewer genes. To further understand the gene content differences between these two genomes, we first clustered their genes into different orthogroups using OrthoFinder software (v2.3.3)27. Among the 55,594 genes, 17,391 pairs were one-to-one orthologs. A total of 14,160 genes (4601 in A. oxysepala var. kansuensis and 9559 in A. coerulea) formed clusters (X:0 clusters, X ≥ 1) with genes from only one of the two species (Fig. 4d). Although in other gene clusters, copy number differences were found between these two species, the aggregate numbers (3579 vs. 3073) were similar (Fig. 4d). Thus, the possession of more X:0 clusters in A. coerulea was the main reason for the interspecies gene number discrepancy. We further conducted a new round of ortholog clustering to identify genes in these X:0 clusters that were likely to be species-specific by including sequences from Amborella trichopoda, Oryza sativa, Vitis vinifera, Arabidopsis thaliana, and Papaver somniferum. This analysis revealed that 2250 and 5119 genes were specific to A. oxysepala var. kansuensis and A. coerulea, respectively.
Moreover, we classified genes into different families to further elucidate the expansion/contraction differences in certain categories between A. oxysepala var. kansuensis and A. coerulea (Fig. 5a). By referring to a database where 13,867 genes in A. coerulea were assigned to 985 families, we were able to put 13,860 genes from A. oxysepala var. kansuensis into 955 families. We found that the two species showed clear differences in the number of genes in some functionally important families (e.g., DUF, F-box, CBM, cytochrome P450, and MADS-box) (Fig. 5b, c and Table S9). For example, this detailed analysis identified 65 and 88 MADS-box genes from A. oxysepala var. kansuensis and A. coerulea, respectively. In line with the contrast evolutionary patterns of Type I (fast birth-and-death) and Type II (highly conserved) MADS-box genes, the two species contained comparable numbers of Type II genes, while A. coerulea contained 18 more Type I genes (Fig. 5c). Because Type II genes play important roles in flower development28,29, we further examined the copy number difference in each subfamily between the two species. In each of the APETALA1 (AP1), SEPALLATA (SEP), APETALA3 (AP3), and SHORT VEGETATIVE PHASE (SVP) subfamilies, A. coerulea contained one more copy. Genes in the first three subfamilies usually determine floral organ identity28, while those in the SVP subfamily control flowering time28. These differences may have impacted the difference in flower traits between A. oxysepala var. kansuensis and A. coerulea; further investigations on these genes would be of interest.

a The number of gene families in each size category and the number of genes from each species. The size of each gene family was defined by referring to the genes in A. coerulea. Note that gene family classifications are not mutually exclusive. b Comparison of gene numbers in the top 15 families (ranked by size). c Phylogenetic tree (neighbor joining, NJ) of MADS-box genes from the two species constructed with the p-distance model (1000 bootstrap replications)
Previous studies have suggested that the entire Chr4 in Aquilegia had a unique evolutionary pattern, with approximately twice the level of polymorphism of the rest of the genome, which might have been caused by reduced purifying selection10. For this reason, we performed two types of analyses. First, we calculated the density of genes on all seven chromosomes (Fig. 6a). We found that the density of genes on A.ox_chr4 was significantly lower than that on the other chromosomes (Benjamini–Hochberg adjusted P ≤ 0.01, one-sided Mann–Whitney U test; Fig. 6b and Table S10). We then calculated the dN/dS ratio of all one-to-one orthologs between A. oxysepala var. kansuensis and A. coerulea. Regardless of the translocation, when comparing genes on A.ox_chr4 with those on the other chromosomes, we found that the dN/dS ratios on this chromosome were the highest, although not every comparison was significant after multiple-testing correction (Fig. 7a and Table S11). This indicates that genes on this chromosome are likely to be evolving under fewer functional constraints.

a Distribution of gene number in each 300 kb window. The arrows on chromosomes 1 and 4 designate rearrangement breakpoints. Pink arrows show that A.ox_chr1_2 is homologous to A.co_chr4_2, while dark-green arrows show the homology of A.ox_chr4_2 with A.co_chr1_2. b Boxplot of gene density on each chromosome. Each point represents the number of genes in a 300 kb window. In the boxplots, each box indicates the lower quartile, median, and upper quartile values. Pairwise significance tests between Chr4 and the other chromosomes within each species were performed. Asterisks indicate significance level: **P < 0.01, ***P < 0.001. Detailed P value for each comparison is provided in Table S10. c Gene density on different homologous fragments. Detailed P value for each comparison is provided in Table S12

a Boxplot of dN/dS values calculated between one-to-one orthologs. Genes were assigned to different chromosomes according to the A. oxysepala var. kansuensis genome. Pairwise significance tests between Chr4 and the other chromosomes were performed. Asterisks indicate significance level: *P < 0.05, **P < 0.01, ***P < 0.001. Detailed P value for each comparison is provided in Table S11. b Boxplot of dN/dS values for one-to-one orthologs distributed on the rearranged chromosome segments. Detailed P value for each comparison is provided in Table S13
However, because there was a rearrangement between A.ox_chr4 and A.ox_chr1, it was not clear whether the entire A.ox_chr4 or just the part that was homologous to A.co_chr4 had this evolutionary pattern. We thus separated the two focal chromosomes into four parts (i.e., A.ox_chr1_1, A.ox_chr1_2, A.ox_chr4_1, and A.ox_chr4_2) according to their homologous relationships with the A. coerulea genome and made further comparisons. We found that A.ox_chr4_1 and A.ox_chr1_2, which were homologous to Chr4 of A. coerulea, had the lowest gene density (Benjamini–Hochberg adjusted P < 0.001, one-sided Mann–Whitney U test; Fig. 6c, Table S12). When comparing dN/dS values, not surprisingly, we found that the highest values were also from genes on A.ox_chr4_1 (median = 0.39) and A.ox_chr1_2 (median = 0.38) (Fig. 7b). However, statistical tests for comparison between A.ox_chr4_1 and other chromosomes/chromosomal segments were significant (Benjamini–Hochberg adjusted P < 0.03, one-sided Mann–Whitney U test; Table S13), while only two of the tests for A.ox_chr1_2 were significant (Benjamini–Hochberg adjusted P < 0.05, one-sided Mann–Whitney U test; Table S13). The discrepancy between these two segments indicates that they may have evolved under different constraints.
Previous studies suggested that Aquilegia species are ancient tetraploids, but the timing of the whole-genome duplication (WGD) event is debated30,31. Guo and colleagues (2018) held that this event occurred after the divergence of Papaveraceae and Ranunculaceae, while Akoz and Nordborg (2019) proposed that it was shared by all eudicots. To better understand this event, we identified all the within-genome syntenic regions of the A. oxysepala var. kansuensis genome and calculated the synonymous substitution rate (dS) of each pair of collinear paralogs using the modified Nei–Gojobori model. For comparison, the dS values of collinear paralogs in A. coerulea and grape and those of the one-to-one orthologs between A. oxysepala var. kansuensis and grape were also estimated. The distributions of dS values for collinear paralogs in the two Aquilegia species peaked at the same position (Fig. S7), confirming that Aquilegia species are ancient tetraploids. Although the grape paralogs had a similar peak value on the distribution plot of dS values, the dS distribution of orthologs of A. oxysepala var. kansuensis and grape peaked at a higher value, indicating that the WGD event occurred after the divergence of Aquilegia and grape. Therefore, our results strongly support that the most recent WGD in Aquilegia occurred after the divergence of Papaveraceae and Ranunculaceae, which is not consistent with the previously hypothesized pre-eudicot tetraploidization30.
Discussion
A high-quality genome of A. oxysepala var. kansuensis
In this study, by combining PacBio, BioNano, and Hi–C data, we have built a nearly complete assembly of the genome of A. oxysepala var. kansuensis. The evaluation results show that the genome is of high quality. In comparison with the A. coerulea genome v3.1, the assembly of A. oxysepala var. kansuensis has better contiguity, with longer contig N50, fewer contigs/scaffolds, and a lower percentage of gaps (Table 1). Notably, the 7 chromosomes of A. oxysepala var. kansuensis were supported by superscaffolds almost as long as chromosome arms. The biggest difference between these two genomes is the total number of protein-coding genes; the A. coerulea genome is annotated with 4452 more genes. Our detailed comparison revealed that a great part of this discrepancy can be attributed to the many more species-specific genes in A. coerulea. Two underlying reasons might account for this. First, these genes were generated de novo or were lost in other species, which may have played important roles in interspecific morphological or physiological divergence. Second, since gene predictions are not error-free, it is possible that some of them have been generated by misannotation. The observation that the mRNA length of the species-specific group was significantly shorter than the genome-wide average (P < 10−199 and P = 0 for A. oxysepala var. kansuensis and A. coerulea, respectively) provides further support for this possibility. Nevertheless, 36.2% (815/2250) of the species-specific genes in A. oxysepala var. kansuensis have Gene Ontology (GO) assignments, compared with only 18.6% (951/5119) in A. coerulea, suggesting that our gene set is of higher confidence. In fact, our annotation is supported by much stronger evidence since we applied both Iso-Seq and RNA-Seq (PE150) data from different tissue samples (Table S8) to annotate the A. oxysepala var. kansuensis genome, in contrast to the annotation of A. coerulea, for which only RNA-Seq (SE35 and SE40) from sepals of this species but numerous sequencing data (mostly SE51) from other species were used. The fact that 97.2% of the genes in A. oxysepala var. kansuensis show homology with functionally annotated genes reaffirms the high confidence of our gene set. All these results show that we obtained a high-quality genome that will pave the way for spur reduction, adaptation, and evolutionary studies of Aquilegia.
Genome divergence between A. oxysepala var. kansuensis and A. coerulea
It is interesting that the two Aquilegia species show considerable differences in their genomes. First, there has been a translocation between chromosomes 1 (A.ox_chr1) and 4 (A.ox_chr4) in A. oxysepala var. kansuensis relative to A. coerulea. Previously, it was observed that Chr4 in two other Aquilegia and one Semiaquilegia species have similar organizational patterns10, suggesting that this chromosome might have a conserved structure. One important piece of evidence is that 5S rDNA loci are uniquely localized to Chr4 in all three species10. We found that in A. oxysepala var. kansuensis, 5S rDNA loci are located on both A.ox_chr1 and A.ox_chr4, the majority of which are on the segments that are homologous to Chr4 of A. coerulea (Aco_chr4) (Table S14). Thus, it is likely that the translocation event was specific to A. oxysepala var. kansuensis. More interestingly, we also found 5S rDNA loci on other segments, including the 2,542,608–2,542,670 and 15,215,141–15,215,260 regions of A.ox_chr1 and the 33,260,556–33,260,618 region of A.ox_chr4 (Table S14). This indicates the possibility that the 5S rDNA was not unique to Aox_chr4 even before the chromosome translocation. Second, the two genomes are different in the number and content of annotated protein-coding genes, transposable elements, and noncoding RNAs. Particularly, among protein-coding genes, only 68.0% (17,391/25,571) of the genes of A. oxysepala var. kansuensis are one-to-one orthologous to genes of A. coerulea, which means that numerous orthologs are different in copy number. Third, it has been reported that the sequence divergence between species of Aquilegia from different geographic regions (such as Asia and North America) is at least 0.81%10. Since a divergence level as low as 0.15% would lead to marked biases in some genetic/genomic analyses32, using either one of the genomes as the reference for the other would be unsatisfactory. These results demonstrate the evolution of chromosomes in the plant genome and further highlight the importance of sequencing the genome of A. oxysepala var. kansuensis.
Special evolutionary pattern of Chr4 of A. oxysepala var. kansuensis
It has been shown that the entire Chr4 of A. coerulea (A.co_chr4) has evolved uniquely under reduced purifying selection, demonstrating a “decaying” nature31 with a much higher level of polymorphism and lower gene density than other chromosomes10. Moreover, it is suggested that this evolutionary pattern of Chr4 began before the split of Aquilegia and Semiaquilegia10. In concordance with this, we found that the chromosome segments in A. oxysepala var. kansuensis (A.ox_chr4_1 and A.ox_chr1_2) that are homologous to A.co_chr4 have a significantly lower gene density. What is surprising is that A.ox_chr4_1 and A.ox_chr1_2 do not show the same pattern in their dN/dS ratios. A.ox_chr4_1 has a significantly higher dN/dS ratio than other chromosomes or chromosome segments, while the difference for A.ox_chr1_2 is weaker. This indicates that the translocation of this segment to Chr1 might have influenced the evolution of genes therein so that they are under more functional constraints than before. In this case, it is tempting to speculate that the translocation of a segment (A.ox_chr4_2) from Chr1, which is likely evolving under high functional constraint (high gene density and low dN/dS ratio), might also have an impact on the “decay” of Chr4. More genomic data from other Aquilegia taxa would be helpful to further clarify this influence.
Materials and methods
Plant material collection, DNA extraction, and sequencing
We excavated whole plants of A. oxysepala var. kansuensis from a population in Yuzhong County, Gansu Province, China (N35°47′33″, E104°3′12″), and cultivated them in a growth chamber. Fresh young leaves of an individual were collected, and genomic DNA was extracted using the TIANGEN DNAsecure Plant Kit (GP1). For short-read sequencing, an ~350 bp insert size pair-end library was constructed and sequenced using the Illumina HiSeq 4000 platform. A total of 18.3 Gb raw data were generated (Table S1). For long-read sequencing, ~20 kb SMRTbell libraries were prepared and sequenced using PacBio Sequel Sequencer, which produced 37.6 Gb raw data (Table S3).
To extract enough DNA for the construction of optical mapping libraries, we collected young, fresh leaves from dark-treated seedlings of A. oxysepala var. kansuensis. High-molecular-weight DNAs were then isolated using the BioNano Prep Plant Tissue DNA Isolation Kit Contents (Part # 80003). Direct Labeling Enzyme 1 (DLE-1) was used to digest the DNAs, which were then fluorescently labeled, stained, and loaded onto a Saphyr Chip for sequencing. Nearly 301 Gb optical mapping data were generated. For the Hi–C data, we constructed two libraries, which were subjected to sequencing on the Illumina HiSeq 4000 platform, yielding a total of 36.3 Gb data (Table S5).
Genome assembly
PacBio SMRT long reads were assembled using Falcon33 (Branch 3.1) (--max_diff 100 --max_cov 100 --min_cov 2 --min_len 5000) after self-correction. The resulting contigs were then polished by Quiver34 using the long reads. SSPACE-LongRead35 was applied to merge the contigs into scaffolds with default parameters. Finally, Illumina reads were mapped back to polish the scaffolds using Pilon (—threads 20 —frags)36.
Genome map assemblies for A. oxysepala var. kansuensis were generated using Bionano Solve Pipeline version 3.3 and Bionano Access version 1.3.0 (https://bionanogenomics.com/support/software-downloads/). Low-quality optical molecules with length ≤ 180 kb or label number ≤9 were removed. A rough assembly was first performed with the following parameters: -i 0 -V 0 -A -z -u –m. A second assembly, using the first round result as reference, was launched with the following parameters: -y -r (rough assembly cmap) -V 0 -m. To create hybrid scaffolds, optical maps were aligned to PacBio-assembled contigs and scaffolded with BioNano’s hybrid-scaffold tool. The process included comparing the BioNano genome nick-based maps to the in silico nick maps of the genome sequence to find their best matches and potential reciprocal scaffolding of each dataset. If there were conflicts between the sequence and optical maps, both of them were cut at the conflict sites and assembled again with the hybrid-scaffold parameter “-B 2 -N 2”.
To construct chromosome-level assemblies, we further applied the 36.3 Gb Hi–C data. Almost 31.9 Gb clean data were retained after removing adapter sequences and low-quality reads, i.e., those with a ratio of N higher than 0.1 and/or quality value less than 5 (Q < 5). Unmapped reads, self-ligated reads, dangling-end reads, internal fragment reads and reads with incorrect sizes were further removed using HiCUP software37. Hi–C reads were mapped to the hybrid scaffolds, and Hi–C contact frequency between genomic loci was computed using Juicer (version 1.7.6)26. 3D-DNA (version 180114)21 was used to anchor and orient scaffolds based on the contact frequency calculated from mapped Hi–C read pairs to obtain the pseudomolecules for two rounds with default parameters. During this process, we manually corrected the misassembled order, oriented scaffolds of DNA based on Hi–C data, and took advantage of the telomere-to-telomere contact enrichment associated with genomes in the Rabl configuration to obtain seven pseudochromosomes using Juicebox Assembly Tools (JBAT version 1.8.8)38. The Hi–C read contact frequency matrix was visualized using Juicebox (version 1.8.8)38.
Genome annotation
Two methods (i.e., homology alignment and de novo annotation) were used to extract the repeats in the genome of A. oxysepala var. kansuensis. The repeats in a plant genome can be divided into tandem repeats and interspersed repeats. For tandem repeats, the software TRF version 4.09 (http://tandem.bu.edu/trf/trf.html) was used to make de novo predictions. For the prediction of transposable elements, two approaches were used: the first was to search against the Repbase database (http://www.girinst.org/repbase) using RepeatMasker (v4.0.5)39 with default parameters, and the second was de novo prediction through LTR_FINDER (v1.0.7) (http://tlife.fudan.edu.cn/ltr_finder/), RepeatScout (v1.0.5) (http://www.repeatmasker.org/), and RepeatModeler (v1.0.3) (http://www.repeatmasker.org/) with default parameters.
Noncoding RNAs (ncRNAs) include tRNA, rRNA, miRNA, and snRNA. tRNA was predicted by tRNAscan-SE40. rRNA was annotated by BLASTN searches against other species. miRNAs and snRNAs were identified by searching against the Rfam database (13.0)41 with default parameters using INFERNAL software (v1.1.2)42.
A combination of three methods, including ab initio prediction, homology-based prediction and transcriptome-assisted prediction, was used to identify protein-coding genes. For homology-based prediction, we used sequences of six species, including A. coerulea, O. sativa, A. trichopoda, and Populus trichocarpa from Phytozome (https://phytozome.jgi.doe.gov/); A. thaliana from TAIR (https://www.arabidopsis.org); and P. somniferum from communications with the relevant authors30. TBLASTN searches (e-value ≤ 1e−5) were then conducted against the A. oxysepala var. kansuensis assembly to identify homologous proteins, which were then aligned to the assembly by GeneWise (v2.4.1)43 to annotate gene structures. For ab initio prediction, we employed Augustus (v3.2.3)44, Geneid (v1.4)45, Genescan (v1.0)46, GlimmerHMM (v3.04)47, and SNAP (v2013.11.29)48 software with default parameters. We used both Iso-Seq and RNA-Seq datasets from different tissue samples (i.e., roots, stems, leaves, flowers, fruits, and seeds) at different stages of development as evidence for gene annotation. We constructed five Iso-Seq libraries, which contained RNAs isolated from five samples, respectively, i.e., leaves of different stages, stems+roots, seedlings, small inflorescences (with flowers of early stages), and large inflorescences (with flowers of medium-late stages and seeds). These libraries were then sequenced on the PacBio Sequel platform, yielding a total of 47.6 Gb raw data. RNA-Seq data were generated by pooling equal amounts of RNA obtained from the above five samples and sequencing on the Illumina HiSeq 4000 platform. RNA-Seq raw reads were filtered and mapped to our genome assembly using TopHat (v2.0.11)49 to identify exon regions and splice positions. The alignment results were then input into Cufflinks (v2.2.1)50 with default parameters for genome-based transcript assembly. Iso-Seq data were processed through the standard Iso-Seq pipeline. The generated transcripts, together with the genome-guided assembly of RNA-Seq data, were integrated with the Program to Assemble Spliced Alignments (PASA)51. A nonredundant reference gene set was generated by merging genes predicted by the three aforementioned methods with EVidenceModeler (EVM, v1.1.1)51 and then updated using PASA51. Functions were assigned to each gene according to its best matches by aligning its protein sequence to the Swiss-Prot and NR databases using BLASTP (e-value ≤ 1e−5). Motifs and domains were annotated using InterProScan70 (v5.31)52 by searching against publicly available databases, including ProDom, PRINTS, Pfam, SMRT, PANTHER, and PROSITE. The GO terms for each gene were assigned according to the corresponding InterPro entry.
Genome alignment and microsynteny detection
We used Nucmer, contained in the MUMmer package version 3.2353, to align the genomes of the two species, with default parameters. The results were then filtered by delta-filter program with “-i 90 -g –q”. To infer gene-level synteny, we used BLASTP (-evalue 1e−20 -num_threads 16 -outfmt 6) to generate protein alignment between the two species. The outputs were then imported into MCScanX54 to identify syntenic blocks.
Orthogroup and gene family classification
OrthoFinder27 (orthofinder -f fastadata -S blast -M msa -T raxml) software was used to group genes of different species, and in-house R scripts were applied to count the number of ortholog clusters obtained. Based on gene family information of A. coerulea (http://www.supfam.org/genome/Ac), we performed BLASTP (-gapopen 11 -gapextend 1 -max_target_seqs 5) searches of each A. oxysepala var. kansuensis sequence against the proteome of A. coerulea and assigned it to a certain gene family based on the best hit.
Phylogenetic analysis of the MADS-box gene family
We first retrieved all MADS-box gene sequences classified by using the gene family information of A. coerulea. When aligning the protein sequences of these genes, we found five in A. coerulea and seven in A. oxysepala var. kansuensis that had incomplete MADS-box domains. We mapped these sequences to their corresponding genomic regions using TBLASTN and manually curated the annotations. All MADS-box protein sequences were aligned using the hmmalign program in the HMMER package version 3.055 and the SRF-domain (MADS-box domain) model downloaded from Pfam (http://pfam.sanger.ac.uk/). The corresponding CDS sequence alignment was generated using PAL2NAL56. Nucleotide sites in the MADS-box domain were used for phylogenetic analysis, which was performed in MEGA757 (pairwise deletion and p-distance model) with 1000 bootstrap replications.
Gene density and dN/dS analyses
Gene density was calculated for nonoverlapping 300 kb windows along the whole chromosome using in-house R scripts. Then the ggplot2 package58 in R version 3.6.2 was used for plotting, and the Wilcox test was employed for statistical analysis. One-to-one ortholog clusters of the two species identified using OrthoFinder were used for dN/dS analysis. Protein sequences of each pair of orthologs were aligned using MUSCLE3.8.3159 software with default parameters. PAL2NAL56 was then applied to create the corresponding CDS sequence alignment, and trimAL software60 was used to remove alignment-ambiguous codons. Finally, the Perl script (dS_dN_MNG.PL) downloaded from the website (https://sites.google.com/view/masafumi-nozawa/scripts) was applied to calculate the dN and dS values using a modified Nei–Gojobori model, and in-house R scripts were used to calculate the dN/dS ratios.
Identification of collinear paralogs and calculation of dS values
We used MCScanX54 software to identify synteny blocks within each of the A. oxysepala var. kansuensis, A. coerulea, and grape genomes. One-to-one orthologs between A. oxysepala var. kansuensis and grape were obtained by using OrthoFinder27. The dS value for each pair of genes was then calculated as described above using the modified Nei–Gojobori model. The geom_density57 function in R was used to compute and draw kernel density estimates of each dS list.
References
Whittall, J. B., Medina-Marino, A., Zimmer, E. A. & Hodges, S. A. Generating single-copy nuclear gene data for a recent adaptive radiation. Mol. Phylogenet. Evol.39, 124–134 (2006).
Kramer, E. M. & Hodges, S. A. Aquilegia as a model system for the evolution and ecology of petals. Philos. Trans. R. Soc. Lond. B365, 477–490 (2010).
Kramer, E. M. Aquilegia: a new model for plant development, ecology, and evolution. Annu. Rev. Plant. Biol.60, 261–277 (2009).
Hodges, S. A. & Derieg, N. J. Adaptive radiations: from field to genomic studies. Proc. Natl Acad. Sci. USA106, 9947–9954 (2009).
Whittall, J. B. & Hodges, S. A. Pollinator shifts drive increasingly long nectar spurs in columbine flowers. Nature447, 706–709 (2007).
Hodges, S. A., Whittall, J. B., Fulton, M. & Yang, J. Y. Genetics of floral traits influencing reproductive isolation between Aquilegia formosa and Aquilegia pubescens. Am. Nat.159, S51–S60 (2002).
Fulton, M. & Hodges, S. A. Floral isolation between Aquilegia formosa and Aquilegia pubescens. Philos. Trans. R. Soc. Lond. B266, 2247–2252 (1999).
Huang, L. et al. Genetic diversity and evolutionary history of four closely related Aquilegia species revealed by 10 nuclear gene fragments. J. Syst. Evol.56, 129–138 (2018).
Xue, C. et al. Morphological variation pattern of Aquilegia ecalcarata and its relatives. J. Syst. Evol.58, 221–233 (2020).
Filiault, D. L. et al. The Aquilegia genome provides insight into adaptive radiation and reveals an extraordinarily polymorphic chromosome with a unique history. Elife7, e36426 (2018).
Fior, S. et al. Spatiotemporal reconstruction of the Aquilegia rapid radiation through next-generation sequencing of rapidly evolving cpDNA regions. N. Phytol.198, 579–592 (2013).
Zhu, R. R., Gao, Y., Zhang, Q. X. & Debener, T. Quantitative trait locus mapping of floral and related traits using an F2 population of Aquilegia. Plant Breed.133, 153–161 (2014).
Yant, L., Collani, S., Puzey, J., Levy, C. & Kramer, E. M. Molecular basis for three-dimensional elaboration of the Aquilegia petal spur. Proc. R. Soc. Lond. B282, 20142778 (2015).
Whittall, J. B., Voelckel, C., Kliebenstein, D. J. & Hodges, S. A. Convergence, constraint and the role of gene expression during adaptive radiation: floral anthocyanins in Aquilegia. Mol. Ecol.15, 4645–4657 (2006).
Voelckel, C., Borevitz, J. O., Kramer, E. M. & Hodges, S. A. Within and between whorls: comparative transcriptional profiling of Aquilegia and Arabidopsis. PLoS ONE5, e9735 (2010).
Sharma, B., Yant, L., Hodges, S. A. & Kramer, E. M. Understanding the development and evolution of novel floral form in. Aquilegia. Curr. Opin. Plant Biol.17, 22–27 (2014).
Puzey, J. R., Gerbode, S. J., Hodges, S. A., Kramer, E. M. & Mahadevan, L. Evolution of spur-length diversity in Aquilegia petals is achieved solely through cell-shape anisotropy. Proc. R. Soc. Lond. B279, 1640–1645 (2012).
Ballerini, E. S., Kramer, E. M. & Hodges, S. A. Comparative transcriptomics of early petal development across four diverse species of Aquilegia reveal few genes consistently associated with nectar spur development. BMC Genomics20, 668 (2019).
Li, L., Luo, Y., Yang, C., Deng, J. & Erst, A. S. Aquilegia yangii (Ranunculaceae), a new species from western China. Phytotaxa348, 289–296 (2018).
Dolezel, J. & Bartos, J. Plant DNA flow cytometry and estimation of nuclear genome size. Ann. Bot.95, 99–110 (2005).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science356, 92–95 (2017).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics26, 589–595 (2010).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics31, 3210–3212 (2015).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics25, 2078–2079 (2009).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res.46, e126 (2018).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst.3, 95–98 (2016).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol.20, 238 (2019).
Smaczniak, C., Immink, R. G. H., Angenent, G. C. & Kaufmann, K. Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development139, 3081–3098 (2012).
Theissen, G., Melzer, R. & Rumpler, F. MADS-domain transcription factors and the floral quartet model of flower development: linking plant development and evolution. Development143, 3259–3271 (2016).
Guo, L. et al. The opium poppy genome and morphinan production. Science362, 343–347 (2018).
Akoz, G. & Nordborg, M. The Aquilegia genome reveals a hybrid origin of core eudicots. Genome Biol.20, 256 (2019).
Nevado, B., Ramos-Onsins, S. E. & Perez‐Enciso, M. Resequencing studies of nonmodel organisms using closely related reference genomes: optimal experimental designs and bioinformatics approaches for population genomics. Mol. Ecol.23, 1764–1779 (2014).
Chin, C. S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods13, 1050–1054 (2016).
Chin, C. S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods10, 563–569 (2013).
Boetzer, M. & Pirovano, W. SSPACE-LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC Bioinform.15, 211 (2014).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE9, e112963 (2014).
Wingett, S. et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Res4, 1310 (2015).
Dudchenko, O. et al. The Juicebox Assembly Tools module facilitates de novo assembly of mammalian genomes with chromosome-length scaffolds for under $1000. Preprint at bioRxiv https://doi.org/10.1101/254797 (2018).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform.Chapter 4, Unit 4.10 (2009).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res.25, 955–964 (1997).
Kalvari, I. et al. Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res.46, D335–D342 (2018).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics29, 2933–2935 (2013).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Res.14, 988–995 (2004).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res.34, W435–W439 (2006).
Alioto, T., Blanco, E., Parra, G. & Guigó, R. Using geneid to identify genes. Curr. Protoc. Bioinform.64, e56 (2018).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol.268, 78–94 (1997).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics20, 2878–2879 (2004).
Korf, I. Gene finding in novel genomes. BMC Bioinform.5, 59 (2004).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics25, 1105–1111 (2009).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc.7, 562–578 (2012).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol.9, R7 (2008).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics30, 1236–1240 (2014).
Marcais, G. et al. MUMmer4: a fast and versatile genome alignment system. PLoS Comput. Biol.14, e1005944 (2018).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res.40, e49 (2012).
Eddy, S. Profile hidden Markov models. Bioinformatics14, 755–763 (1998).
Suyama, M., Torrents, D. & Bork, P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res.34, W609–W612 (2006).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol.33, 1870–1874 (2016).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis. (Springer, 2016).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res.32, 1792–1797 (2004).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics25, 1972–1973 (2009).
Acknowledgements
We thank Scott Hodges, Yi Ren, and members of the Kong laboratory for valuable discussions; Fangdong Geng for providing the pictures of A. oxysepala var. kansuensis; Cheng Xue and Li Sun for their help in the collection of field samples; and Zhe Cai for technical assistance with the server. This work was supported by grants from the National Natural Science Foundation of China (Nos. 31930008, 31870207, and 31570227), the CAS/SAFEA International Partnership Program for Creative Research Teams, and the National Ten Thousand Talents Program of China.
Author information
Authors and Affiliations
Contributions
H.K. and G.X. conceived and supervised the study. Y. Jiang, R.Z., J.X., and M.W. collected the samples. R.Z. and X.G. extracted the RNA. J.X. and K.L. assembled the genome and assessed genome quality with the help of Y. Jiao. M.W. and X.G. checked the RNA-seq data. J.X. carried out comparative genomics analysis with the assistance of H.Z. B.Y., J.X., and G.X. performed the phylogenetic analysis. H.Z. calculated the gene densities and dN/dS ratios, and conducted statistical analyses. H.Z. and G.X. provided R scripts for plotting. J.X., H.K., Y. Jiao, and G.X. wrote the paper. All authors read, edited, and approved the final paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xie, J., Zhao, H., Li, K. et al. A chromosome-scale reference genome of Aquilegia oxysepala var. kansuensis. Hortic Res 7, 113 (2020). https://doi.org/10.1038/s41438-020-0328-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-020-0328-y
This article is cited by
-
Identification of errors in draft genome assemblies at single-nucleotide resolution for quality assessment and improvement
Nature Communications (2023)
-
Analysis of the Coptis chinensis genome reveals the diversification of protoberberine-type alkaloids
Nature Communications (2021)
-
The chromosome-level reference genome of Coptis chinensis provides insights into genomic evolution and berberine biosynthesis
Horticulture Research (2021)
