
Abstract
Cytosolic Ca2+ plays a key role in signal transduction in plants. Calcium imaging is the most common approach to studying dynamic changes in the cytoplasmic Ca2+ content. Here, we used mature ‘Fuji’ apples (Malus pumila Mill.) to obtain viable protoplasts from flesh tissue cells by enzymatic hydrolysis; then, three small-molecule fluorescent probes (fluo-8/AM, fluo-4/AM, and rhod-2/AM) were loaded into the protoplasts. All three Ca2+ fluorescent probes successfully entered the cytoplasm but did not enter the vacuole. Both the Ca2+ chelator (EGTA) and Ca2+ channel blocker (La3+) reduced the fluorescence intensity in the cytoplasm. The calcium ionophore A23187 increased the fluorescence intensity in the cytoplasm at 5 µmol/L but decreased it at 50 µmol/L. Additionally, A23187 reversed the fluorescence intensity in the cytoplasm, which was decreased by La3+. Ionomycin is also a calcium ionophore that can increase the fluorescence intensity of calcium in the cytoplasm. These results suggest that small-molecule Ca2+ fluorescent probes can be used to detect changes in cytosolic calcium levels in the cells of fruit flesh tissue. In addition, the optimum concentration of fluo-8/AM was determined to be 5 µmol/L. This was the first time that protoplasts have been isolated from apple flesh tissue cells and small-molecule fluorescent probes have been used to detect calcium in the cytoplasm of flesh tissue cells. This study provides a new method to study calcium signal transduction in fruit flesh tissue.
Similar content being viewed by others


Introduction
Calcium influences many fruit qualities1,2; it affects fruit firmness, sugar content, storage period, and physiological disorders during storage3,4. Thus, a low calcium content reduces fruit firmness and shortens the length of the storage period. A disorder in cell calcium metabolism can cause apple bitter pit5, brown spot disease6, and cock spot in Pyrus serotina7. In vegetable crops, tomato umbilical rot is also caused by a calcium metabolism disorder8. These physiological disorders severely degrade fruit quality and cause serious economic losses to growers2,9. Therefore, growers often supplement fruit with calcium. Spraying calcium early during fruit growth and soaking in calcium at postharvest can improve fruit firmness10, reduce the incidence of bitter pit11 and cock spot7, delay fruit senescence and softening, and extend the storage period2.
However, calcium supplementation is not always effective. Studies have shown that calcium spraying or soaking does not always increase fruit firmness3. Spraying calcium on the fruit skin surface is not an effective way to prevent litchi fruit cracking12, nor can it reduce the incidence of apple bitter pit, as was shown in a study in which calcium spraying was repeated five times over the entire apple fruit growth period13. Moreover, other studies have shown that the calcium content of bitter-pit fruit is not lower than that of healthy fruit3,14. The main reason for these conflicting results is that the role of calcium in flesh tissue cells is not clear15. In the cytoplasm, Ca2+ acts as a signaling ion that mediates a variety of cell growth and development processes16.
Calcium imaging has been demonstrated to be a powerful method for observing the dynamic changes in intracellular Ca2+ in living cells with good spatial and temporal resolution17,18. At present, there are two methods for intracellular calcium imaging in living cells: one uses small-molecule fluorescent probes, and the other uses FRET (Förster Resonance Energy Transfer)—based genetically encoded sensors (GECIs)19. Although GECIs have many advantages, a stable transgenic system for fruit trees is difficult to establish, and the growth period of fruit trees is inconveniently long;20 furthermore, the weak fluorescence of single cells is not conducive to overcoming background noise, and the method is susceptible to interference from endogenous calmodulin and other shortcomings21.
Small-molecule fluorescent indicators, such as fluo-4/AM and fluo-8/AM, show Ca2+-specific selectivity and are noninvasively loaded by esterification incubation, which is flexible, rapid, and not cytotoxic22. Fluo-4/AM was successfully loaded into the pollen tube of Pyrus pyrifolia20 and petunia23 as well as into the guard cells24 and root hairs25,26 of Arabidopsis. Fluo-8/AM can also be used to detect dynamic calcium in guard cells27.
At present, the study of plant calcium imaging mainly focuses on pollen tubes, root hairs, and guard cells. There are few reports on calcium fluorescence staining of flesh cells. Calcium may be the most important mineral determining the quality of fruits, especially apples and pears, because they are commonly stored for long periods of time28. Moreover, apples are economically important worldwide and a healthy food29. Today, ‘Fuji’ apples are one of the most popular sweet apple varieties in the world and are commercially grown in Japan, China, the United States, and Australia. Especially in China, ‘Fuji’ is the main planting variety, and its yield and cultivated area account for more than 60% of the total apple production and total cultivated area30. Here, we first obtained viable protoplasts from ‘Fuji’ apple flesh tissue cells and then detected Ca2+ in the cytoplasm with a small-molecule calcium fluorescent reagent.
Results
Flesh tissue staining with Ca2+ fluorescence
Apple fruit flesh was manually cut as thin as possible with a surgical knife. The slices did not display fluorescence prior to loading with fluo-8/AM (Fig. 1a), which indicated that the flesh tissue cells were not self-fluorescing. A cryostat was then used to cut the flesh tissue to a thickness of 80 µm (i.e., a single cell layer), and then the tissue was stained with fluo-8/AM at 37 °C for 30 min. In this case, fluorescence was observed only around the cell, i.e., against the cell wall (Fig. 1b). Although the manually cut flesh tissue was sliced as thin as possible, it was difficult to ensure that the slices were single-cell layers. Nonetheless, after loading fluo-8/AM into the slices, the staining results from the hand-sliced tissue were consistent with those from the slices cut with the cryostat and only showed fluorescence at the cell edges (Fig. 1c). Then, single flesh cells were obtained by enzymatic hydrolysis. Despite some fluorescence at the edge of the cells after fluo-8/AM loading, the fluorescence intensity was very low (Fig. 1d). As there is a large vacuole in the center of the flesh cell with the cytoplasm squeezed around the cell (Supplementary Fig. S1A,B), fluorescence appeared around the cell. The cell wall is a pool of Ca2+ that can also be combined with a Ca2+ fluorescence indicator;31 thus, it was difficult to determine whether fluorescence was from the cell wall or from the cytoplasm.

a Slice of flesh tissue that was not loaded with fluo-8/AM. b Flesh tissue was cut by a cryostat and then fluorescently stained with fluo-8/AM. c Flesh tissue was cut by hand and then fluorescently stained with fluo-8/AM. d Fluo-8/AM loaded into single flesh cells
Protoplast viability assay
We used an enzymatic method to obtain protoplasts from individual apple flesh cells (Fig. 2a and Methods). Some protoplasts had vacuoles, while others did not (Fig. 2b). We measured the diameter of 50 protoplasts and found an average diameter of 48 µm (Supplementary Fig. S2). Protoplasts were stained with FDA for 5 min and showed fluorescence in the cytoplasm, indicating that the isolated protoplasts were viable (Fig. 2c). Protoplasts were stored at 37 °C for 30 min and then stained with FDA; they were still fluorescent (Fig. 2d), indicating that the high temperature (37 °C) did not affect their viability.

a (1) A healthy and mature apple was selected from which a piece was cut; (2) a sample of the mesocarp was cut; (3) the mesocarp cube was cut to 10 × 5 × 1 mm3 (4) mesocarp cubes were placed in a centrifuge tube containing the enzyme solution. b Protoplasts of different diameters. Arrowheads point to the protoplast, and arrows point to the vacuole in the protoplast. c Protoplasts were stained with FDA. d Protoplasts were incubated at 37 °C for 30 min and then stained with FDA
Protoplast calcium ion fluorescence staining
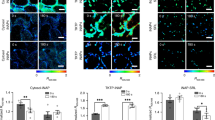
We loaded different small fluorescent indicators into the protoplasts (Methods) to measure cytoplasmic Ca2+. Protoplasts showed no fluorescence when no Ca2+ fluorescent indicator was loaded into them (Fig. 3a). On the other hand, when either fluo-8/AM or fluo-4/AM were loaded into the protoplasts, the cytoplasm was fluorescent, but the vacuole was not (Fig. 3b, c). Furthermore, when rhod-2/AM was loaded into the protoplast, there was still fluorescence in the cytoplasm and none in the vacuoles (Fig. 3d). However, the results of rhod-2 staining were different from those of fluo-4/AM or fluo-8/AM staining. The fluorescence in the protoplasts was granular (Fig. 3d), as rhod-2/AM entered the mitochondria and stained Ca2+ within the mitochondria32. These results suggested that fluo-4/AM and fluo-8/AM successfully stained Ca2+ in the cytoplasm and that there was no compartmentalization in either case33.

a Intact protoplast without any loaded fluorescent probes. b Protoplast loaded with fluo-8/AM. c Protoplast loaded with fluo-4/AM. d Protoplast loaded with rhod-2/AM
Effects of different concentrations of fluo-8/AM on the fluorescence intensity of calcium in protoplasts
We used fluo-8/AM as a reference reagent to detect the effect of different concentrations of a fluorescent reagent on the fluorescence intensity of calcium in the cytoplasm. When the concentration of fluo-8/AM increased from 0.1 to 5 μmol/L, the fluorescence intensity of calcium ions gradually increased (Fig. 4a–d). Particularly from 1 to 5 μmol/L, the fluorescence intensity increased significantly (P < 0.001) (Fig. 4g). However, when fluo-8/AM exceeded 5 μmol/L, the fluorescence intensity of calcium decreased (Fig. 4e, f), but the difference was not significant (p > 0.05) (Fig. 4g). We calculated the diameters of protoplasts after different concentrations of fluo-8/AM staining. When the concentration of fluo-8/AM exceeded 5 μmol/L, the diameter of the protoplasts decreased (Supplementary Fig. S3), mainly due to the increase in the ratio of protoplast shrinkage or breakage (Supplementary Fig. S4). Although there are no specific reports on the effect of fluo-8/AM on protoplast membranes, we believe that the high concentration of fluo-8/AM had a destructive effect on the protoplast membrane. Therefore, we suggest that the optimal concentration of fluo-8/AM for calcium fluorescence staining in protoplasts of flesh cells is 5 μmol/L.

a Control protoplast not loaded with fluo-8/AM. b 0.5 µmol/L. c 1 µmol/L. d 5 µmol/L. e 25 µmol/L. f 50 µmol/L. g Statistical analysis of fluorescence intensity in the protoplasts. As there was no self-fluorescence phenomenon in the control, there was no statistical control of fluorescence density. *** Indicates a significant difference (P < 0.001, Student’s t-test). Vertical bars indicate ± SE. Each data point represents the mean of 10 protoplasts. a–f Representative fluorescence images of protoplasts
Reagents that affect the response of Ca2+ fluorescence in protoplasts
A Ca2+ chelator (EGTA)34 was added to the protoplasts with fluo-8/AM at final concentrations of 1 mmol/L or 10 mmol/L. EGTA is a chelating agent for Ca2+ that can significantly reduce fluorescence intensity (Fig. 5a, b). Additionally, La3+, which is a calcium-ion channel blocker on the cell membrane35, was added to protoplasts during fluo-8/AM loading; La3+ could also significantly decrease fluorescence intensity, regardless of whether the final concentration was 10 or 100 µmol/L (Fig. 5c, d). Protoplasts were treated with calcium ionophore A2318736,37 to a final concentration of 5 µmol/L when fluo-8/AM was loaded into the protoplasts. This treatment might have increased the fluorescence intensity in the cytoplasm relative to that of the controls (Fig. 6a, b, g); however, contrary to expectation, A23187 significantly decreased fluorescence intensity when it reached a final concentration of 50 µmol/L (Fig. 6a, c, g), likely owing to La3+-mediated reduction of fluorescence intensity (Fig. 6d–g). We observed changes in Ca2+ fluorescence in the same protoplast. The protoplasts without any treatment (control) showed a slight decrease in fluorescence intensity within 25 min (Fig. 7a), and the change was not significant. La3+ and EGTA reduced the Ca2+ fluorescence intensity within 5 min and completely quenched the fluorescence within 25 min (Fig. 7b, c). A23187 increased the Ca2+ fluorescence intensity in protoplasts within 5 min, but the fluorescence intensity reached a peak at 10 min and then decreased slightly (Fig. 7d). We also used another calcium ionophore (ionomycin) to increase the fluorescence intensity of the protoplasts, and the fluorescence intensity did not decay within 25 min (Fig. 7e). In addition, after La3+ reduced the fluorescence of the same protoplast, supplementation with A23187 increased the calcium fluorescence intensity (Supplementary Fig. S5). This result further demonstrated that fluo-8/AM can stain Ca2+ in the cytoplasm and show dynamic changes in the Ca2+ content. Calcium in the cytoplasm is maintained by an influx of extracellular calcium.

a Protoplast after loading with fluo-8/AM, followed by the addition of 1 mmol/L EGTA, b 10 mmol/L EGTA, c 10 µmol/L La3+, or d 100 µmol/L La3+

a Control protoplast loaded with fluo-8/AM. b Protoplast after loading with fluo-8/AM, followed by the addition of 5 µmol/L A23187 or c 50 µmol/L A23187; d protoplast after loading with fluo-8/AM, followed by the addition of 100 µmol/L La3+. e Protoplast after loading with fluo-8/AM, followed by the addition of 5 µmol/L A23187 and 100 µmol/L La3+. f 50 µmol/L A23187 and 100 µmol/L La3+. g Statistical analysis of fluorescence intensity in the protoplasts. *** Indicates a significant difference (P < 0.001, Student’s t-test). * Indicates a significant difference (P < 0.05, Student’s t-test). Vertical bars indicate ± SE. Each data point represents the mean of 10 protoplasts. a–f Representative fluorescence images of protoplasts

a Protoplast loaded with fluo-8/AM (Control). b The calcium ion fluorescence was observed for 5 min, and then 100 µmol/L La3+ was added. c The calcium ion fluorescence was observed for 5 min, and then 10 mmol/L EGTA was added. d Calcium ion fluorescence was observed for 5 min, and then 5 µmol/L A23187 was added. e The calcium ion fluorescence was observed for 5 min, and then 1 µmol/L ionomycin was added. Vertical bars indicate ± SE. Each data point represents the mean of three protoplasts. a–e Representative fluorescence images of protoplasts
Discussion
Calcium imaging is a useful technique for studying the roles of Ca2+ in living cells18. In plants for which stable transgenic systems are difficult to establish, small-molecule calcium fluorescent indicators are commonly used for cellular calcium imaging20. We used enzymatic hydrolysis to obtain viable apple flesh protoplasts and then loaded Ca2+ fluorescent probes into the protoplasts for cytoplasmic calcium imaging. Ca2+ in the cytoplasm plays an important role in signaling38. Under CO2 and high pressure, strawberry firmness was shown to increase, but it was suggested that such an increase would be delayed if intracellular Ca2+ efflux was inhibited39. Therefore, calcium imaging in the cytoplasm of flesh cells provides a powerful tool for studying the physiological role of calcium in fruit growth and development40.
Small-molecule calcium fluorescent probes cannot cross the membrane into the cell. Therefore, the indicator should be added with an acetoxymethyl (AM) ester, and the dye should be made neutral so that it can cross the cell membrane41. However, esterases on the cell membrane can cleave AM groups and prevent the indicator from entering the cell20. There are a variety of methods for loading fluorescent probes into plant cells. Zhang et al. (1988)42 loaded fluo-3/AM at a low temperature (4 °C) so that it would enter the root tip cells. Similarly, Qu et al. (2012)43 used fluo-3/AM, but at a high temperature (37 °C), to image the inside of pollen tubes. These methods aimed to reduce the activity of esterases on the cell membrane. To overcome the barrier of the cell membrane, the microinjection method has been used to directly inject the fluorescent probes into the cell. The cell wall is a pool of Ca2+ that interferes with the fluorescent intensity of Ca2+ in the cytoplasm when loaded with a fluorescent probe31. To avoid this interference, the microinjection method is also used to avoid the cell wall44. However, this method is very complicated and slow31,33. In a previous study, we used cell lysates to slightly degrade the cell membrane without reducing cell viability and allowed the calcium fluorescent probe to enter the pollen tube20. In the present study, we successfully removed the cell wall of flesh cells by enzymatic hydrolysis and loaded fluorescent probes into protoplasts at a high temperature (37 °C). This method did not affect the viability of the protoplasts. Additionally, we loaded fluo-8/AM into the protoplasts at low temperature (4 °C) but failed to stain cytoplasmic Ca2+ (Supplementary Fig. S5).
Using small-molecule calcium fluorescent probes to stain intracellular calcium is not as advantageous as using GECIs. Indeed, a major drawback of the former method is that once the probe enters the cell, it is subject to compartmentalization33. In other words, it is easy for the probe to accumulate in the vacuole. In the cytoplasm, calcium concentrations are only 100–200 nmol/L, which is much lower than the concentration in the vacuole45, where it ranges from 0.1 to 10 mmol/L45. Nonetheless, the vacuole showed almost no fluorescence in this study, a finding that suggests that none of the three calcium fluorescent probes used were compartmentalized in the protoplasts. In addition, calcium fluorescence was uniformly distributed in protoplasts without vacuoles (Supplementary Fig. S6).
Intracellular Ca2+ fluorescence intensity can be reduced by lowering the Ca2+ concentration46. In the present study, the Ca2+ chelator EGTA seriously decreased the fluorescence intensity of calcium. In turn, La3+ blocks calcium channels on the cell membrane; however, the results of La3+ treatment in this study regarding the effects of cytoplasmic calcium were inconsistent. It has been suggested that a significant reduction in cytoplasmic Ca2+ concentration might be due to the inhibition of extracellular calcium influx47. However, some studies suggest that although La3+ inhibits extracellular calcium influx, it can also cause stored intracellular calcium to be released, thereby increasing Ca2+ cytoplasmic concentration48. In the experiments reported herein, La3+ significantly reduced Ca2+ concentration in the cytoplasm of the flesh cells, and calcium ionophore A23187 reversed the inhibitory effect of La3+ in vitro.
Babcock et al. (1976) studied the effects of A23187 on Ca2+ in bovine epididymal spermatozoa. They suggested that the effects of A23187 on the intracellular Ca2+ concentration were highly complex36. A23187 promotes Ca2+ efflux at low concentrations, while it promotes intracellular accumulation of Ca2+ at high concentrations36. However, 0.1 µmol/L A23187 inhibited the absorption of Ca2+ in the cardiac sarcoplasmic reticulum and only promoted it at 1 nmol/L49. Other studies have suggested that the effects of A23187 on Ca2+ flux depend on extracellular Ca2+ concentration50. Thus, for example, A23187 increased the Ca2+ concentration in hepatic stellate cells at 30 µmol/L but caused cell apoptosis51. Consistently, in the present study, A23187 increased the Ca2+ concentration in the cytoplasm at low concentrations (5 µmol/L) but decreased it at high concentrations (50 µmol/L). As a calcium carrier52, it has been reported that ionomycin can increase the concentration of calcium in the cytoplasm53, and in this study, ionomycin increased the fluorescence density of calcium in the cytoplasm of apple pulp cells.
Postharvest softening of apples is a very serious problem for apple growers in many countries4. Softening of apple fruits is generally considered an undesirable ripening process because firmer apples tend to have more juice and are crisper crunchier and less mealy than softer apples54. Ca2+ plays a key role in fruit firmness14. Therefore, calcium is added to the fruit before or after harvesting to increase the firmness of the fruit or reduce the speed of fruit softening55. We supplemented Ca2+ and EGTA through the fruit stalk 15 days before the harvest of ‘Golden Del. Reinders’ apple. Exogenous Ca2+ could increase the firmness of the fruit, while EGTA reduced the firmness of the fruit (Supplementary Fig. S8A). Exogenous Ca2+ also increased the cytosolic calcium concentration of fruit cells, while EGTA decreased the cytosolic concentration (Supplementary Fig. S8B,C). The results suggested that there was a positive correlation between Ca2+ concentration in the cytoplasm and fruit firmness. The rapid physiological degradation after harvest greatly reduced the quality and marketability of cassava (Manihot esculenta Crantz). Exogenous Ca2+ reduces postharvest physiological deterioration by increasing endogenous levels of Ca2+ and inducing the expression of genes related to melatonin biosynthesis after harvest. These effects are reversed by the exogenous application of EGTA56. Our findings support this result. However, Deell et al. (2001) suggested that the application of Ca2+ has nothing to do with the firmness of apples3. In addition, the firmness of cherry fruits regulated by exogenous Ca2+ is the same as that of apples regulated by exogenous Ca2+, which is also contradictory15. This is because the response of the fruit to Ca2+ is still unknown. Therefore, the application of Ca2+ imaging technology to study the relationship between the dynamic changes of Ca2+ in the cytoplasm and the physiological activities of pulp cells will help us to explore the regulatory role of Ca2+ in fruit firmness, soluble solids content, and physiological diseases in fruits, such as bitter pit.
In conclusion, we obtained viable protoplasts by enzymatic hydrolysis and then successfully loaded three small-molecule probes into the protoplasts at a high temperature (37 °C) for 30 min. The fluo-8/AM and fluo-4/AM fluorescence intensity was uniformly distributed in the cytoplasm of protoplasts and can be used at 5 μmol/L (Optimal Concentration) to determine the calcium concentration in the cytoplasm. In turn, rhod-2 was granulated in the cytoplasm and can be used to study calcium in the mitochondria. This method can provide technical support for calcium research in fruit and vegetable flesh tissue cells.
Methods
Production of flesh slices
We selected disease-free and mature ‘Fuji’ (Malus domestica Borkh. CV. Fuji) apples. The flesh tissue at 1–2 cm under the exocarp (skin) was cut with a scalpel. The flesh tissue was precooled in an embedding solution57, and 80 µm thick slices were cut with a microtome cryostat (Leica CM3050 S, Nussloch, Germany).
Protocol for the extraction of single cells from apple flesh tissue
In accordance with our previously published method58, the flesh 1–2 cm below the exocarp was cut into small pieces of 1 × 1 × 1 mm3 and placed in a CPW (cell protoplast washing)59 solution containing 0.1% of macerozyme R-10 (w/v) at 28 °C and centrifuged at 70 rpm for 30 min in the dark. The enzyme solution was washed three times with CPW, stirred for 1 h with a magnetic stirrer, and centrifuged at 800 rpm for 3 min; the pulp with single cells was collected.
Protoplast extraction protocols
The following basic solution was prepared to extract protoplasts: 20 mmol/L CaCl2·2H2O, 5 mmol/L MES, 0.6 mol/L D-sorbitol, and Tris buffer. The solution was adjusted to pH 5.8. The enzymatic solution was prepared from the following basic solution: 0.004 mg/ml macerozyme R-10 (Yakult, Japan), 0.0001 mg/ml pectolase Y-23 (Yakult, Japan), and 0.002% mg/ml cellulase R-10 (Yakult, Japan). The flesh tissue under the exocarp was cut into small pieces that were 10 × 5 × 1 mm3 in size (Fig. 2a), which were placed into 1.5 ml centrifuge tubes and added to 0.5 ml of the enzymatic solution. After the enzyme solution was digested at 28 °C for 1.5 h, it was immediately placed on ice to stop the reaction and then washed three times with a basic solution by centrifugation at 1000 rpm. Finally, the protoplast suspension was obtained.
Protoplast viability assay
FDA (Fluorescein Diacetate, Thermo Fisher, USA) was dissolved in DMSO (Dimethyl Sulfoxide) to produce a 1 mg/ml stock solution. One microliter of stock solution was added to 99 μL of DMSO to prepare a working solution. Then, 99 μL of protoplast suspension was placed into a 0.2 ml centrifuge tube, and 1 μL of the FDA working solution was added. The staining was carried out for 5 min at 25 °C in the dark. Before observation, the stained sediments of protoplast suspensions were washed three times with basic solution by centrifugation at 1000 rpm. Then, the viability of the protoplasts was tested under a fluorescence microscope (EVOS Auto 2, Thermo Fisher, USA). We selected the light cube of GFP because the excitation wavelength of FDA is 490 nm60.
Calcium ion fluorescence staining
Fluorescent loading solutions were prepared based on our previously published methods43. The concentration of the loading solution for preparing different kinds of fluorescent indicators was 0.5 mmol/L; a volume of 99 μL of sliced flesh tissue, single cell or protoplast suspension was placed into 0.5 ml centrifuge tubes, and 1 μL of a loading solution of fluo-4/AM, fluo-8/AM or rhod-2/AM (Dojindo Laboratories, Kumamoto, Japan) was added to make the final concentration of the fluorescent dye 5 µmol/L. We loaded the fluorescent dye into the cells for 30 min at 37 °C in the dark. After loading, the dye was washed three times with a basic solution to remove excess fluorescent dye and observed with a fluorescence microscope. Since the excitation wavelength of fluo-4/AM and fluo-8/AM is 490 nm, we selected GFP as the light cube. In turn, RFP was used as the light cube when loading rhod-2/AM because the excitation wavelength of rhod-2/AM is 551 nm.
Measuring fluorescent trace
We took 18 μL of protoplasts after loading with fluo-8/AM, dropped it onto an adhesive on a concave microscope slide, covered the slide with a coverslip, and observed the calcium fluorescence change in the cells with a fluorescence microscope for 5 min. We then added 2 μL of EGTA (Ethylene Glycol-bis (beta-aminoethyl ether) -N,N,N′,N′-Tetraacetic Acid), A23187, ionomycin, or La3+ reagents so that their final concentrations were 10 mmol/L, 5 μmol/L, 1 μmol/L or 100 μmol/L, respectively. We then continued to observe the changes in calcium fluorescence for 25 min and took photographs every 5 min.
Image analysis
The fluorescence results were analyzed using Image-Pro Plus 6.0 software (Media Cybernetics, Inc., MD, USA) according to our published methods20. For final processing, we used Adobe Photoshop CS5 (Adobe Systems, Mountain View, CA).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7.0 software (GraphPad Software, Inc., La Jolla, CA, USA). Data are expressed as the means ± SD. Student’s t-test was used to analyze the differences among the experimental groups.
References
Barrett, D. M., Beaulieu, J. C. & Shewfelt, R. L. Color, flavor, texture, and nutritional quality of fresh-cut fruits and vegetables: desirable levels, instrumental and sensory measurement, and the effects of processing. Crit. Rev. Food Sci. Nutr. 50, 369–389 (2010).
Gao, Q., Xiong, T., Li, X., Chen, W. & Zhu, X. Calcium and calcium sensors in fruit development and ripening. Sci. Horticulturae 253, 412–421 (2019).
Deell, J. R., Khanizadeh, S., Saad, F. & Ferree, D. C. Factors affecting apple fruit firmness–a review. J.- Am. Pomological Soc. 55, 8–27 (2001).
Johnston, J., Hewett, E. & Hertog, M. A. T. M. Postharvest softening of apple (Malus domestica) fruit: a review. N.Z. J. Exp. Agriculture 30, 145–160 (2002).
Miqueloto, A. et al. Mechanisms regulating fruit calcium content and susceptibility to bitter pit in cultivars of apple. Acta horticulturae 1194, 469–474 (2018).
Kou, X. et al. Effects of CaCl2 dipping and pullulan coating on the development of brown spot on ‘Huangguan’ pears during cold storage. Postharvest Biol. Technol. 99, 63–72 (2015).
Raese, J. T. & Drake, S. R. Effects of preharvest calcium sprays on apple and pear quality. J. Plant Nutr. 16, 1807–1819 (1993).
Vinh, T. D. et al. Comparativeanalysis on blossom-end rot incidence in two tomato cultivars in relation to calcium nutrition and fruit growth. Horticulture J. 87, 97–105 (2018).
Yamane, T. Foliar calcium applications for controlling fruit disorders and storage life in deciduous fruit trees. Jpn. Agric. Res. Q. 48, 29–33 (2014).
Wojcik, P. Yield and “jonagold” apple fruit quality as influenced by spring sprays with commercial rosatop material containing calcium and boron. J. Plant Nutr. 25, 999–1010 (2002).
Miqueloto, A., Amarante, C. V. T. D., Steffens, C. A., Santos, A. D. & Mitcham, E. J. Relationship between xylem functionality, calcium content and the incidence of bitter pit in apple fruit. Sci. Horticulturae 165, 319–323 (2014).
Huang, X.-M. et al. Spraying calcium is not an effective way to increase structural calcium in litchi pericarp. Sci. Horticulturae 117, 39–44 (2008).
Ernani, P. R., Dias, J., Amarante, C. V. T. D., Ribeiro, D. C. & Rogeri, D. A. Preharvest calcium sprays were not always needed to improve quality of ‘gala’ apples in Brazil. Rev. Brasileira De. Frutic. 30, 892–896 (2008).
Hocking, B., Tyerman, S. D., Burton, R. A. & Gilliham, M. Fruit calcium: transport and physiology. Front. Plant Sci. 7, 569–569 (2016).
Winkler, A. & Knoche, M. Calcium and the physiology of sweet cherries: a review. Sci. Horticulturae 245, 107–115 (2019).
Demidchik, V., Shabala, S., Isayenkov, S., Cuin, T. A. & Pottosin, I. Calcium transport across plant membranes: mechanisms and functions. N. Phytologist 220, 49–69 (2018).
Bootman, M. D., Rietdorf, K., Collins, T. J., Walker, S. & Sanderson, M. J. Ca2+-sensitive fluorescent dyes and intracellular Ca2+ imaging. CSH Protoc. 2013, 83 (2013).
Grienberger, C. & Konnerth, A. Imaging Calcium in Neurons. Neuron 73, 862–885 (2012).
Krebs, M. et al. FRET-based genetically encoded sensors allow high-resolution live cell imaging of Ca2+ dynamics. Plant J. 69, 181–192 (2012).
Qu, H., Xing, W., Wu, F. & Wang, Y. Rapid and inexpensive method of loading fluorescent dye into pollen tubes and root hairs. PLoS ONE 11, e0152320 (2016).
Kanchiswamy, C., Malnoy, M., Occhipinti, A. & Maffei, M. Calcium imaging perspectives in plants. Int. J. Mol. Sci. 15, 3842–3859 (2014).
Gee, K. R. et al. Chemical and physiological characterization of fluo-4 Ca2+-indicator dyes. Cell Calcium 27, 97–106 (2000).
Suwińska, A., Wasąg, P., Zakrzewski, P., Lenartowska, M. & Lenartowski, R. Calreticulin is required for calcium homeostasis and proper pollen tube tip growth in Petunia. Planta 245, 909–926 (2017).
Sun, L. et al. NADK2 positively modulates abscisic acid-induced stomatal closure by affecting accumulation of H2O2, Ca2+ and nitric oxide in Arabidopsis guard cells. Plant Sci. 262, 81–90 (2017).
Niu, Y. et al. Magnesium availability regulates the development of root hairs in Arabidopsis thaliana (L.) H eynh. Plant, cell Environ. 37, 2795–2813 (2014).
Wang, Y. et al. Disruption of actin filaments induces mitochondrial Ca2+ release to the cytoplasm and [Ca2+]c changes in Arabidopsis root hairs. BMC Plant Biol. 10, 53 (2010).
Jing, X., Cai, C., Fan, S., Wang, L. & Zeng, X. Spatial and temporal calcium signaling and its physiological effects in Moso bamboo under drought. Stress. For. 10, 224 (2019).
Faust, M. Physiology of Temperate Zone Fruit Trees. (John Wiley & Sons, Inc., 1989).
Boyer, J. & Liu, R. H. Apple phytochemicals and their health benefits. Nutr. J. 3, 5 (2004).
Shara Aranoff, C. et al. (ed UNITED STATES INTERNATIONAL TRADE COMMISSION) (Publication ITS-04, Washington, DC 20436, 2010).
Gilroy, S., Bethke, P. C. & Jones, R. L. Calcium homeostasis in plants. J. Cell Sci. 106, 453–461 (1993).
Jeanquartier, C. et al. Studying mitochondrial Ca2+ uptake – A revisit. Mol. Cell. Endocrinol. 353, 114–127 (2012).
Takahashi, A., Camacho, P., Lechleiter, J. D. & Herman, B. Measurement of intracellular calcium. Physiological Rev. 79, 1089–1125 (1999).
Schwartz, A. Role of Ca and EGTA on stomatal movements in Commelina communis L. Plant Physiol. 79, 1003–1005 (1985).
Qu, H., Shang, Z., Zhang, S., Liu, L. & Wu, J. Identification of hyperpolarization‐activated calcium channels in apical pollen tubes of Pyrus pyrifolia. N. Phytologist 174, 524–536 (2007).
Babcock, D. F., First, N. L. & Lardy, H. A. Action of ionophore A23187 at the cellular level. Separation of effects at the plasma and mitochondrial membranes. J. Biol. Chem. 251, 3881–3886 (1976).
Verma, A. et al. Calcium ionophore A23187 reveals calcium related cellular stress as “I-Bodies”: an old actor in a new role. Cell calcium 50, 510–522 (2011).
Edel, K. H., Marchadier, E., Brownlee, C., Kudla, J. & Hetherington, A. M. The evolution of calcium-based signalling in plants. Curr. Biol. 27, R667–R679 (2017).
Hua, W. M. et al. Potential role of pectate lyase and Ca2+ in the increase in strawberry fruit firmness induced by short-term treatment with high-pressure CO2. J. Food Sci. 79, S685–S692 (2014).
Russell, J. T. Imaging calcium signals in vivo: a powerful tool in physiology and pharmacology. Br. J. Pharmacol. 163, 1605–1625 (2011).
Richter, P. & Häder, D. In IMAGE ANALYSIS: Methods and Applications (2nd) (eds Donat-P. Häder & Dr. rer. nat) 373–389 (CRC Press, 2000).
Zhang, W., Rengel, Z. & Kuo, J. Determination of intracellular Ca2+ in cells of intact wheat roots: loading of acetoxymethyl ester of Fluo‐3 under low temperature. Plant J. 15, 147–151 (1998).
Qu, H., Jiang, X., Shi, Z., Liu, L. & Zhang, S. Fast loading ester fluorescent Ca2+ and pH indicators into pollen of Pyrus pyrifolia. J. Plant Res. 125, 185–195 (2012).
Gilroy, S., Fricker, M. D., Read, N. D. & Trewavas, A. Role of calcium in signal transduction of commelina guard cells. Plant Cell 3, 333–344 (1991).
Bose, J., Pottosin, I. I., Shabala, S. S., Palmgren, M. G. & Shabala, S. Calcium efflux systems in stress signaling and adaptation in plants. Front. Plant Sci. 2, 85 (2011).
Palmer, A. E. & Tsien, R. Y. Measuring calcium signaling using genetically targetable fluorescent indicators. Nat. Protoc. 1, 1057–1065 (2006).
Li, Z., Zhang, Z., Yu, M., Zhou, Y. & Zhao, Y. Effects of lanthanum on calcium and magnesium contents and cytoplasmic streaming of internodal cells of Chara corallina. Biol. Trace Elem. Res. 143, 555–561 (2011).
Gelli, A. & Blumwald, E. Calcium retrieval from vacuolar pools (characterization of a vacuolar calcium channel). Plant Physiol. 102, 1139–1146 (1993).
Murray, J. J., Kuzmin, A. V., Reed, P. W. & Levitsky, D. O. Low concentrations of A23187 increase calcium uptake by cardiac sarcoplasmic reticulum. Am. J. Physiol. 249, 1211–1215 (1985).
Reinhart, P. H., Taylor, W. M. & Bygrave, F. L. The effect of ionophore A23187 on calcium ion fluxes and α-adrenergic-agonist action in perfused rat liver. Biochem. J. 214, 405–412 (1983).
Li, Y. et al. Effects of calcium Ionophore A23187 on the apoptosis of hepatic stellate cells stimulated by transforming growth factor-β1. Cell. Mol. Biol. Lett. 23, 1 (2018).
Hao, H. et al. Clathrin and membrane microdomains cooperatively regulate RbohD dynamics and activity in Arabidopsis. Plant Cell 26, 1729–1745 (2014).
Kaya, H. et al. Ca2+-activated reactive oxygen species production by Arabidopsis RbohH and RbohJ is essential for proper pollen tube tip growth. Plant Cell 26, 1069–1080 (2014).
Kolniak-Ostek, J., Wojdyło, A., Markowski, J. & Siucińska, K. 1-Methylcyclopropene postharvest treatment and their effect on apple quality during long-term storage time. Eur. Food Res. Technol. 239, 603–612 (2014).
Jideani, A. I. O., Anyasi, T. A., Mchau, G. R. A., Udoro, E. O. & Onipe, O. O. In Postharvest Handling (ed Ibrahim Kahramanogl) (IntechOpen, 2017).
Hu, W. et al. Crosstalk between calcium and melatonin affects postharvest physiological deterioration and quality loss in cassava. Postharvest Biol. Technol. 140, 42–49 (2018).
Peters, S., Fitzgerald, R., Green, S. & Nunnciato, C. T. Paper Cryoembedding. J. Histotechnol. 26, 173–178 (2003).
Guan, Y. & Haiyong, Q. U. A rapid method for isolating single cells from apple flesh. Horticultural Plant J. 3, 4–9 (2017).
Ning, J., Yali, Z. & Fang, X. An efficient protocol for model legume root protoplast isolation and transformation. Front. Plant Sci. 9, 670 (2018).
Proffitt, R. T., Tran, J. V. & Reynolds, P. A fluorescence digital image microscopy system for quantifying relative cell numbers in tissue culture plates. Cytometry 24, 204–213 (1996).
Acknowledgements
The work was supported by the National Key Research and Development Plan Project: Integrated research and demonstration on the technology of reducing application and increasing efficiency of chemical fertilizer and pesticide in apple cultivation (2016YFD0201120).
Author information
Authors and Affiliations
Contributions
Q.H.Y. conceived and designed the study. Q.L.N. performed the experiments. Q.L.N. and W.Y.Z. analyzed the data. Q.H.Y. wrote the paper. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qiu, L., Wang, Y. & Qu, H. Loading calcium fluorescent probes into protoplasts to detect calcium in the flesh tissue cells of Malus domestica. Hortic Res 7, 91 (2020). https://doi.org/10.1038/s41438-020-0315-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-020-0315-3
