
Abstract
Bitter gourd (Momordica charantia) is a popular cultivated vegetable in Asian and African countries. To reveal the characteristics of the genomic structure, evolutionary trajectory, and genetic basis underlying the domestication of bitter gourd, we performed whole-genome sequencing of the cultivar Dali-11 and the wild small-fruited line TR and resequencing of 187 bitter gourd germplasms from 16 countries. The major gene clusters (Bi clusters) for the biosynthesis of cucurbitane triterpenoids, which confer a bitter taste, are highly conserved in cucumber, melon, and watermelon. Comparative analysis among cucurbit genomes revealed that the Bi cluster involved in cucurbitane triterpenoid biosynthesis is absent in bitter gourd. Phylogenetic analysis revealed that the TR group, including 21 bitter gourd germplasms, may belong to a new species or subspecies independent from M. charantia. Furthermore, we found that the remaining 166 M. charantia germplasms are geographically differentiated, and we identified 710, 412, and 290 candidate domestication genes in the South Asia, Southeast Asia, and China populations, respectively. This study provides new insights into bitter gourd genetic diversity and domestication and will facilitate the future genomics-enabled improvement of bitter gourd.
Similar content being viewed by others


Introduction
Bitter gourd (Momordica charantia) is an economically important vegetable crop in the family Cucurbitaceae, which also includes common vegetables and fruits such as cucumber (Cucumis sativus), watermelon (Citrullus lanatus), and melon (Cucumis melo). Bitter gourd is native to Africa1 but was domesticated in Asia over a long period of time, with written Sanskrit records dating back to Indo-Aryan culture (2000 to 200 BC)2. M. charantia var. muricata (small fruited; hereafter, muricata) was first identified by Willdenow in the Hortus Malabaricus3,4, a book from the seventeenth century describing the flora of southern India, where other researchers later inferred it to be the wild progenitor of cultivated M. charantia5,6,7. However, the evolutionary trajectory and genetic basis underlying the domestication of bitter gourd remain largely unknown8.
Bitter gourd is a popular vegetable characterized by its bitter fruits. This bitterness is a result of cucurbitane triterpenoids, including cucurbitacins (sapogenins) and cucurbitane glycosides (saponins)9,10. Bitter gourd is often used in folk medicine to manage type 2 diabetes, and recent clinical studies have confirmed its role in lowering elevated fasting glucose levels in prediabetes patients11,12,13.
Although bitter gourd has been cultivated for centuries, the improvement of its varieties and cultivars has been hindered by the extreme genetic homogeneity of commercial varieties, as well as the low-genetic diversity in natural populations14; therefore, there is great demand for genetic resources that can improve bitter gourd varieties.
Recently, a draft genome sequence of the bitter gourd line Momordica charantia OHB3-1 was reported, with a scaffold-level genome assembly of 285.5 Mb and 45,859 protein-coding genes annotated by ab initio prediction15. However, a more accurately annotated chromosome-level genome assembly for bitter gourd is still necessary. Population-scale genomic variation analysis by resequencing has been shown to be a powerful approach for revealing the genetic diversity and genetic basis underlying domestication in many crops, including rice16, maize17, soybean18, cucumber19, and tomato20. However, to our knowledge, no studies have investigated population-scale genomic variation in bitter gourd.
Here, we report high-quality genome sequences for bitter gourd. In addition, we resequenced 187 bitter gourd germplasms from a worldwide collection, as well as one M. balsamina and one M. foetida accession. Our data provide an improved understanding of bitter gourd diversity and domestication, paving the way for efficiently breeding new bitter gourd cultivars.
Materials and methods
Sample collection and genome sequencing
De novo whole-genome sequencing was conducted in two bitter gourd lines, M. charantia Dali-11 collected from Foshan city, Guangdong Province, China, and the small-fruited line TR collected from Singida, Tanzania. For Dali-11, libraries with an increasing gradient of insert sizes of 170 bp, 500 bp, 800 bp, 2 kb, 5 kb, 10 kb, and 20 kb were constructed and sequenced on the Illumina HiSeq 2000 platform. Nine paired-end libraries were generated, and 12 lanes were sequenced, producing 92.46 Gb of raw data. Low-quality reads, including short-insert library reads comprising 40% of bases with quality scores ≤7 and large-insert library reads that comprising >35% of bases with quality scores ≤7, were filtered out, as were PCR duplicates in which read1 and read2 of two paired-end reads were completely identical. The filtration of low-quality and duplicated reads resulted in 75.31 Gb (~251× coverage) of data for genome assembly. For TR, six paired-end libraries with insert sizes of 270 bp, 800 bp, 2 kb, 5 kb, and 10 kb were prepared. In total, ~70.55 Gb and 55.68 Gb (~185× coverage) of raw data and clean data were generated, respectively, for subsequent genome assembly.
The samples that were resequenced were as follows: 166 M. charantia (including 136 intermediate-size- to large-fruited M. charantia and 30 small-fruited muricata) samples, 21 small-fruited TR-group samples, one M. balsamina line, and one M. foetida line (Supplementary Table S27). Sequencing libraries were constructed according to the manufacturer’s instructions (Illumina). Short reads were generated by applying the SolexaPipeline-0.3 base-calling pipeline (Illumina). Approximately 10–38x coverage of the genome sequences from each sample was generated.
Genome assembly
After correction and filtering for short-read sequences, the bitter gourd genomes were assembled using SOAPdenovo21. Contigs were constructed using paired-end reads of short-insert-size libraries, and the contigs were connected using long-insert-size libraries to generate scaffolds. All reads were used to fill gaps in the scaffolds. To assemble the Dali-11 genome, scaffolds were anchored to pseudochromosomes through a high-quality RAD genetic map22.
The quality and completeness of our assemblies were assessed according to the following methods. First, all clean reads were mapped to the corresponding assembly to investigate the completeness of the assemblies, which can be reflected by the mapping ratio obtained using SOAP223 with default parameters, and SOAPcoverage 2.27 (http://soap.genomics.org.cn/) was then used to calculate sequencing depth. Second, we searched for conserved genes by using BUSCO24 (http://busco.ezlab.org/) software. In addition, we de novo assembled the transcriptome data of six tissues, including roots, stems, leaves, male flowers, ovaries, and fruits, into unigenes and then mapped them back to the bitter gourd genomes.
Transcriptome analysis
To generate transcriptomes, total RNA was extracted from Dali-11 roots, stems, leaves, male flowers, ovaries, and fruits from four developmental stages (6, 12, 18, and 24 days after pollination) using TRIzol reagent (Invitrogen, Carlsbad, CA) following the manufacturer’s instructions. The raw transcriptome reads containing adaptors or >10% unknown nucleotides and those showing low quality (>50% bases with a quality value ≤5) were filtered, and the clean reads were then mapped to the Dali-11 reference gene using Bowtie225 and to the genome using TopHat26. The expression level for individual genes was quantified according to fragments per kilobase of exon per million reads mapped (FPKM) values using RSEM27.
Genome annotation
Repetitive sequences in the bitter gourd genomes were identified using a combination of TRF28, Repbase-based29,30 and de novo methods. Three de novo analysis programs, including LTR-FINDER31, PILER32, and RepeatScout33, were used to generate the initial repeat library. Then, the de novo library was analyzed using RepeatMasker to annotate and classify repeats.
For gene annotation, we used homology, ab initio prediction, and transcript data to predict gene structure in the Dali-11 genome. The homology approach involved mapping protein sequences from three other cucurbit species (C. lanatus, C. sativus, and C. melo) to the Dali-11 genome using TBLASTN34 (E-value < 1e-5), and the homologous genome sequences were aligned against the matching proteins using GeneWise35. TopHat and Cufflinks were used to obtain transcript structures from RNA-seq data from the various tissues and developmental stages. Augustus (augustus-3.0.3) was employed for ab initio gene prediction36. GLEAN37 was used to merge the results from the homology and transcript analysis to form a comprehensive and nonredundant reference gene set. The genes in the TR genome were predicted using the homology-based and de novo methods described above. Once gene structures were identified in Dali-11 and TR, gene functions were assigned based on the best alignment attained using BLASTP against the Nr, SWISS-PROT38, TrEMBL38, GO39, KEGG40, and InterProScan41 databases.
Genome evolution analysis
The distribution of orthologous gene families in M. charantia (Dali-11), C. lanatus, C. melo, C. sativus, C. pepo, C. maxima, C. moschata, L. siceraria, and J. regia was defined using OrthoMCL42. The resulting 2248 shared single-copy genes were used to generate the phylogeny of M. charantia (Dali-11) and the eight other plant species. Divergence time estimations between species were determined using MCMCtree in PAML (v4.5)43. The divergence time of ~84 Mya between Fagales and Cucurbitales indicated by fossil information44, as well as two calibrated divergence times, 26.28 Mya between C. moschata and C. lanatus45 and 10.10 Mya between C. melo and C. sativus46, were used to estimate the divergence time in this study.
Paralogous genes were detected using the all-versus-all BLASTp method (E-value < 1e-5), and homologous blocks were detected using MCScanX47. Fourfold degenerate sites (4DTv) values were calculated on the basis of concatenated nucleotide alignments with HKY substitution models48.
SNP and InDel detection
Paired-end reads (clean reads) were mapped to the Dali-11 and TR genomes using BWA49, which resulted in a BAM file. SAMtools Picard and GATK50,51 were used for further handling procedures such as alignment, repeat removal, and ID addition. The GATK pipeline was used to detect SNPs and InDels for each sample. Small insertions and deletions (≤50 bp in length) were identified in this study.
Population analysis
Three SNP matrixes (including two separated and one combined SNP set called from the genomes of Dali-11 and TR) were used to construct neighbor-joining phylogenetic trees with PHYLIP 3.69 (http://evolution.genetics.washington.edu/phylip.html). Bootstrap values were calculated with VCF2Dis software (https://github.com/BGI-shenzhen/VCF2Dis). Principal component analysis (PCA) was performed using the EIGENSTRAT stratification correction method52, and the population structure was estimated using FRAPPE53 with calculated K values ranging from two to five.
The correlation coefficient (r2) of alleles was calculated to measure the level of linkage disequilibrium (LD) using PopLDdecay (https://github.com/BGI-shenzhen/PopLDdecay). The LD blocks were analyzed with Haploview54.
The genetic separation between individual genomes was inferred via the multiple sequentially Markovian coalescent (MSMC) method55, with a generation time of 1 year and a rate of 1.0 × 10-8 mutations per nucleotide per generation56. We also measured nucleotide diversity (θπ), Watterson’s estimator (θw)57, Tajima’s D58, and Wright’s fixation index (FST)59 in or between different bitter gourd populations according to the corresponding formulas.
To identify the regions underlying the genetic changes caused by different geographic areas of domestication, 30 wild (muricata) samples (Wild30) and groups of 30 large-fruited bitter gourd samples from South Asia (SA30), Southeast Asia (SEA30), and China (CHIN30) were selected. The diversity ratios and cross-population composite likelihood ratios (XP-CLR)60 between SA30, SEA30, and CHN30 and Wild30 were calculated, and regions were identified as domestication regions when both the π (θπ) values and XP-CLR ratios were in the top 5% of the distribution outliers.
Results
Sequencing and de novo assembly of the bitter gourd genomes
We performed whole-genome sequencing of the bitter gourd cultivar Dali-11 (M. charantia) from Guangdong, China, and the wild small-fruited line TR from Singida, Tanzania (Supplementary Figs. S1 and S2). Both lines had an estimated genome size of approximately 300 Mb, which was lower than the 339 Mb genome size of OHB3-1 and that of other cucurbits (Table 1 and Supplementary Figs. S3 and S4, Supplementary Table S1). After filtering, we generated a total of 75.3 Gb (251.0×) and 55.7 Gb (185.0×) of high-quality genomic reads for Dali-11 and TR, respectively (Supplementary Tables S2 and S3). The resulting de novo assembly contained 293.6 and 296.3 Mb scaffolds for Dali-11 and TR, with N50 lengths of 3.3 and 0.6 Mb, respectively (Table 1 and Supplementary Tables S4 and S5). We mapped all clean reads back to the Dali-11 assembly. The mapping ratios of all short- and large-insert-size libraries were 94.80% and 82.65%, respectively (Supplementary Table S6), and the assembly contained 96.2% of the 59,740 unigenes assembled from the transcriptome sequences of various tissues (Supplementary Tables S7 and S8). As a new draft genome, the Dali-11 assembly exhibited more complete BUSCOs (96.7%) than the OHB3-1 assembly (95.8%) (Supplementary Table S9). Using the newly developed RAD genetic map22, a total of 113 scaffolds (~90% of the scaffolds were larger than 1 Mb), covering ~85.5% (251.3 Mb) of the Dali-11 assembly (Supplementary Figs. S5 and S6), were anchored to 11 pseudochromosomes (MC01 to MC11). Among the 113 scaffolds, 80 were oriented by at least two markers (Supplementary Table S10).
Repeat sequence and protein-coding gene annotation
We found that ~41.5% (121.8 Mb) and 39.9% (118.2 Mb) of the Dali-11 and TR assemblies consisted of transposable elements (TEs), among which, 31.8% and 33.1% were long-terminal repeat (LTR) retrotransposons (Table 1 and Supplementary Tables S11–S14). The bitter gourd genome has apparently accumulated more LTR retrotransposons over the past 4 million years compared to the cucumber, watermelon, and melon genomes (Supplementary Fig. S7). To facilitate gene annotation, we generated ~537 million clean transcriptome reads from the roots, stems, leaves, flowers, and fruit tissue of Dali-11 (Supplementary Table S7). Using an integrated method (transcriptome, homology-based, and ab initio approaches), we predicted 26,427 high-confidence protein-coding genes in the Dali-11 genome (Supplementary Tables S15 and S16). For the TR genome, we annotated 28,827 protein-coding genes by using the homology-based and ab initio approaches (Supplementary Tables S17 and S18). The number of genes predicted in both bitter gourd genomes was close to that in the cucumber, watermelon, and melon genomes but much lower than that in the OHB3-1 genome (Table 1). The comparative analysis of gene completeness showed that the complete BUSCO ratio of M. charantia Dali-11 (95.9%) and TR (95.5%) was higher than those of M. charantia OHB3-1 (82.20%), C. lanatus (86.50%), C. melo (86.9%), Cucurbita pepo (92.8%), C. sativus (94.8%), and Lagenaria siceraria (88.2%) but comparable to those of Cucurbita maxima (95.70%) and Cucurbita moschata (95.8%) (Supplementary Table S9). Approximately 85.2% and 85.5% of the predicted Dali-11 and TR genes, respectively, were functionally annotated (Supplementary Tables S19 and S20).
Genome comparison within the Cucurbitaceae family
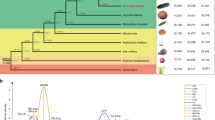
In total, 2248 single-copy orthologous genes were identified in cucumber (C. sativus)61, melon (C. melo)62, watermelon (C. lanatus)63, bitter gourd (M. charantia), zucchini (C. pepo)64, pumpkin (C. maxima and C. moschata)45, bottle gourd (L. siceraria)65, and walnut (Juglans regia)66 (Fig. 1a). Phylogeny and molecular clock analysis based on the 2248 shared single-copy genes indicated that according to our species sampling, M. charantia split from the distantly related genus Cucurbita approximately 36.5 million years ago (Mya) (Fig. 1a), indicating that it is an older species compared to other cucurbit crops67. Similar to cucumber61, melon62, and watermelon63, no recent whole-genome duplication (WGD) has occurred in the M. charantia genome based on the distribution of 4DTv (Fig. 1b). Via genome synteny analysis, we identified 992, 807, and 922 large syntenic blocks, and these syntenic regions contained 14,938, 14,567, and 14,804 genes in C. lanatus, C. melo, and C. sativus, respectively (Fig. 1c and Supplementary Table S21). Moreover, we identified 22,507 gene families (ORTHOMCL clusters) in bitter gourd and eight other plant species (Supplementary Tables S22–S24), and 468 gene families containing 2,071 genes were unique to the bitter gourd genome (Supplementary Tables S25 and S26). With the exception of the annotated genes that were particularly overrepresented in the pathways of oxidative phosphorylation (ko00190), starch and sucrose metabolism (ko00500), and plant-pathogen interaction (ko04626), most of the unique bitter gourd genes had unknown functions.

a Phylogeny and divergence times of nine plant species (M. charantia, C. lanatus, C. melo, C. sativus, C. pepo, C. maxima, C. moschata, L. siceraria, and J. regia) and clusters of orthologous and paralogous gene families in the nine plant species identified by OrthoMCL. b The 4DTv distribution of duplicate gene pairs in M. charantia, C. pepo, C. sativus, and V. vinifera. c Syntenic blocks between the M. charantia genome and the three other cucurbit genomes, where syntenic blocks are defined as regions with >20 genes
Phylogenetic analysis identifies a new species or subspecies of bitter gourd
We resequenced 189 Momordica accessions selected from a panel of 212 accessions collected from around the world68 (Fig. 2a and Supplementary Table S27). Among these accessions, one M. balsamina and one M. foetida sample were designated as the outgroup. We generated ~8.2 billion clean paired-end reads (~1.0 trillion base pairs of sequences), with an average GC ratio of 37.0% and Q20 of 92.1% (Supplementary Table S27). After aligning these clean reads to the Dali-11 genome, the mapping rate ranged from 88.2% to 98.8%, and the average depth ranged from 8.8 to 37.8 among different samples (Supplementary Table S28). Furthermore, we identified a total of 14,450,193 SNPs and 1,588,578 InDels (shorter than 50 bp; Supplementary Tables S29 and S30). Next, we aligned the clean reads to the TR genome and identified 12,170,007 SNPs and 1,572,660 InDels (Supplementary Tables S31–S33). To analyze the evolutionary history of bitter gourd, we conducted a phylogenetic analysis using the separated and combined whole-genome SNPs called from the Dali-11 and TR assemblies and rooted the tree with M. foetida. Interestingly, we found that the 21 small-fruited samples showing a similar morphology to TR formed a distinct clade (Fig. 2b and Supplementary Figs. S8 and S9, Supplementary Tables S34–S36), suggesting that they are a distinct monophyletic group (temporarily designated the TR group) that originated independently.

a The geographic distribution of the 187 bitter gourd samples included in the population analysis. b Neighbor-joining tree for 189 Momordica samples with different taxa represented by different colors: TR (red), muricata (yellow), charantia (blue), and M. balsamina and M. foetida (black) as outgroups. c Phylogeny of Momordica with the inferred divergence time between TR and M. charantia (Supplementary Fig. S10). d MSMC plots of inferred genetic divergence times (colored lines) among TR, muricata, and charantia. The split between the two genomes is defined as the time when the cross-coalescence rate drops to 50%
The divergence time between the M. charantia and TR group was estimated to be ~1.9 Mya (Fig. 2c and Supplementary Fig. S10), which is much longer than the history of human domestication (~10,000 years)69. We then conducted multiple sequential Markovian coalescent (MSMC) analysis with the Dali-11 and TR genome sequences and THMC155 (muricata) resequencing data. The results showed that Dali-11 diverged from TR >200,000 years ago, and that Dali-11 diverged from THMC155 ~6000 years ago (Fig. 2d). We further identified 6,595,112 SNPs in the 166 M. charantia samples and 6,098,414 SNPs in the 21 TR-group samples (Table 2 and Supplementary Table S37). The nucleotide diversity values (θπ, θw) for the TR group samples (θπ = 6.59 × 10−3, θw = 5.37 × 10−3) were significantly higher than those for the M. charantia population (θπ = 1.76 × 10−3, θw = 3.88× 10−3; Table 2 and Supplementary Fig. S11). The fixation index value (FST) between the two populations reached 0.85 (Supplementary Fig. S11 and Supplementary Table S38). We found that TR group samples (Tajima’s D = 0.79) may lack rare alleles or that the group may be under balancing selection. In comparison, the M. charantia population (Tajima’s D = −1.71) may harbor an excess of rare alleles or have recently undergone a population expansion (Table 2)70. Moreover, M. charantia exhibited a higher average dN/dS ratio in genic regions (1.24) compared to the TR group (1.05; Table 2). We found considerable differences between the genetic diversity of the TR group and M. charantia populations. The TR group diverged from M. charantia before human domestication. Overall, these results suggest that the TR group may be a new species or subspecies independent of M. charantia.
Geographic diversity of M. charantia
Based on a neighbor-joining tree, we found that all 30 samples of muricata were nested within the cultivated M. charantia clade and that many were basal to a cluster of M. charantia (Fig. 2b), supporting the aforementioned conclusion that muricata is the wild progenitor of M. charantia. In addition, the 166 M. charantia germplasms can be separated into four geographically differentiated gene pools: South Asia, Southeast Asia, China, and Tanzania (Fig. 3a). This geographic division was illustrated by both population stratification and principal component analysis (PCA) (Fig. 3b, c and Supplementary Table S39). The germplasms from South Asia and China were more differentiated, and the germplasms from Southeast Asia were relatively mixed and exhibited genetic heterogeneity (Fig. 3b, c). In total, we identified 3,311,877, 5,036,642, 1,764,264, and 880,137 SNPs in the South Asia, Southeast Asia, China, and Tanzania groups, respectively (Table 2). The South Asia group exhibited the highest genetic diversity, with a θπ value of 1.94 × 10−3 (Table 2). Tajima’s D in the South Asia group (–0.79) was higher than those in the Southeast Asia (–1.61) and China groups (–1.45), suggesting lower genetic diversity in the last two groups (Table 2). The different geographic groups exhibited variable LD decay values, among which the Southeast Asian population presented the highest (42.6 kb), followed by the Chinese (1.5 kb) and South Asian (0.7 kb) populations (Fig. 3d). These findings support the hypothesis that the domestication of M. charantia in Asia was driven in South Asia71. Furthermore, we found that the 30 muricata samples were distributed across South Asia, Southeast Asia, and China, suggesting either that wild and cultivated bitter gourd dispersed together or that there were multiple domestication events. To identify the genetic regions under selection, we selected 30 large-fruited M. charantia samples from each of South Asia, Southeast Asia, and China (the selected groups are referred to as SA30, SEA30, and CHN30, respectively) and calculated the diversity ratios and the XP-CLR values between the geographic groups and the muricata population (Wild30). Combining the top 5% of θπ and XP-CLR outliers, we identified 6854, 9794, and 7052 selected regions in SA30, SEA30, and CHN30 populations, comprising 710, 412, and 290 genes, respectively (Fig. 3e and Supplementary Figs. S12–S14, Supplementary Tables S40 and S41). Many of these domestication genes were enriched in various metabolic processes (Supplementary Tables S42–S44). These candidate domestication genes will provide the foundation for the identification of associations with key domestication traits.

a The neighbor-joining tree of 166 M. charantia samples. b Model-based clustering analysis with cluster numbers of two to five. c Principal component analysis of M. charantia from different regions. d Decay of LD measured by r2 in the four geographic groups. e Whole-genome screening of domestication regions in the populations of CHN30, SA30, and SEA30. SA South Asia, SEA Southeast Asia, CHN China, TZ Tanzania. The horizontal red dashed lines indicate the threshold defining the top 5% of π values (above the lines) and XP-CLR scores (below the lines)
Comparative analysis of cucurbitane triterpenoid biosynthesis genes
Cucurbitane triterpenoids are the major bitter substances in various cucurbit vegetables, and they are synthesized through the mevalonate pathway72. Cucurbitadienol synthase (CPQ)73 catalyzes the conversion of 2,3-oxidosqualene (2,3-OS) to cucurbitadienol, resulting in the basic skeletal structure of cucurbitane triterpenoid (Fig. 4a). Then, the cucurbitadienol skeleton is further modified by tailoring enzymes, mainly cytochrome P450s (P450s) and UDP-glycosyltransferases (UGTs), to produce diverse cucurbitane triterpenoids. The CPQ orthologs in cucumber and watermelon are Bi74 and CcCDS2/cla007080, respectively75. We identified MC07g0002 as the closest homolog of CPQ in bitter gourd, and the function of this gene in cyclizing 2,3-oxidosqualene to generate cucurbitadienol in yeast has recently been validated76. The CPQ phylogenetic tree showed that MC07g0002 clustered with Siraitia grosvenorii CPQ (SgCPQ), forming a group separated from the orthologs in C. pepo, C. sativus, C. melo, and C. lanatus (Supplementary Fig. S15). We found that MC07g0002 expression was not tissue specific and that the expression level was positively correlated with the bitterness of the tissues, including different developmental stages of fruit (Fig. 4b). Other cucurbits have a conserved Bi cluster responsible for the biosynthesis of cucurbitacin C, B, and E in the same genomic region74,77. We used MC07g0002 as bait to search for other coexpressed genes with predicted functions. Interestingly, the main putative cucurbitane triterpenoid biosynthesis genes, including McCPQ (MC07g0002), two P450s (MC02g_new0213 and MC06g1647), and two UGTs (MC04g0771 and MC01g0394), were not genetically linked to McCPQ in the bitter gourd genome (Fig. 4c and Supplementary Figs. S16–S19). This is similar to the mogroside pathway genes found in the S. grosvenorii genome78.

a The cucurbitane triterpenoid biosynthesis pathway in bitter gourd; the red text denotes the enzymes along with the corresponding predicted enzyme-encoding genes (blue). b Heat map of gene expression for cucurbitane triterpenoid biosynthesis candidate genes. Log2-scaled fragments per kilobase per million (FPKM) values are shown, ranging from low (blue) to high (red) expression. Fruit-6, Fruit-12, Fruit-18, and Fruit-24 represent four developmental stages (6, 12, 18, and 24 days after pollination). The “bitterness” bar indicates that the bitter gourd root, stem, leaf, male flower, ovary, fruit-6, and fruit-12 tissues are bitter, while bitterness decreases from fruit-18 to fruit-24. c The distribution of Bi clusters in cucurbits. Bi clusters are conserved in C. sativus, C. melo, and C. lanatus but lost in M. charantia. The conserved gene clusters span regions of 30–69 kb consisting of six genes, purple: OSCs; blue: P450s; gold: ACTs; gray: unrelated genes. d Distribution of nucleotide diversity (π) at the MC06g_new0561 (McBt1) and MC06g2002 (McBt2) loci in four populations of bitter gourd. SA South Asia, SEA Southeast Asia, CHN China
Two homologs of the bHLH transcription factors Bl and Bt regulate the expression of cucurbitane triterpenoid biosynthesis in cucumber leaves and fruits74, respectively. We identified MC06g_new0561 and MC06g2002 as orthologs of these genes in bitter gourd (Supplementary Fig. S20 and Supplementary Table S45). Both MC06g_new0561 (McBt1) and MC06g2002 (McBt2) were moderately expressed from the ovary to the fruit-12 period and showed weaker expression from the fruit-18 to fruit-24 periods; their expression pattern was highly similar to the expression of MC07g0002 in bitter gourd fruit (Fig. 4b). We also found that MC06g2002 exhibited a higher expression level in bitter gourd roots compared with MC06g_new0561 (Fig. 4b). As a result of domestication, the other three cucurbits, cucumber, melon, and watermelon, underwent a convergent reduction in the genetic diversity of Bl and Bt77. Interestingly, we did not observe obvious reductions in the genetic diversity of McBt1 and McBt2 in the three cultivated geographic populations of bitter gourd, suggesting that there may be a weak signature of artificial selection in bitter gourd around genes regulating bitterness (Fig. 4d). In addition, we found that the region of McCPQ (MC07g0002) presented the same haplotype in 90.6% of the M. charantia samples (including both wild and cultivated bitter gourds) (Supplementary Fig. S21). Thus, bitterness has not been intensively selected among modern bitter gourd cultivars.
Discussion
Bitter gourd is widely consumed in many Asian countries and is used in dietary interventions for diabetes. Here, we report high-quality genome sequences for bitter gourd. Compared to a previous ab initio prediction of genes using the OHB3-1 line15, we provide a more confident gene set using the Dali-11 genome. Bitter gourd represents an early-branching clade of the family Cucurbitaceae45,79, and the genome sequences offer an opportunity to investigate the genomic and biological characteristics of early cucurbits.
By resequencing diverse bitter gourd samples, we gained valuable insights into the genetic diversity, taxonomy, and domestication of bitter gourd. In addition, we provide further evidence that southern Asia is a domestication center of bitter gourd. The differentiation of the 21 TR group samples from M. charantia was firmly supported by our molecular phylogenetic analyses, which were consistent with fruit and seed morphology. We deduced that the 21 TR accessions may belong to a new species or subspecies independent of M. charantia, or the results could support the previously reported M. charantia ssp. macroloba80. The genetic variation between the two species or subspecies can contribute to the utilization of bitter gourd germplasm resources through inter-specific or inter-subspecific crosses to yield improved cultivars in the future. Our findings regarding the geographic diversity and domestication of M. charantia lay the groundwork for future genetic improvement in bitter gourd.
In particular, the Bi gene cluster, which regulates cucurbitane triterpenoid biosynthesis in other popular cucurbit crops, appears to have evolved after the divergence of bitter gourd. The clustering of genes at the Bi locus leads to co-inheritance, co-expression and co-regulation of genes81,82 and may have been driven by intense selection, possibly making this an important locus for rapid responses to stresses83. The lack of co-inheritance of biosynthetic genes and the weak selection for regulatory genes indicate that cucurbitane triterpenoids may play a different role in the response to environments, which may also underlie the bitterness of the bitter gourd fruit. Different tailoring genes, such as P450s and UGTs, can influence the properties of the final structures of cucurbitane triterpenoids77,81, which may also contribute to the bitterness of bitter gourd compared with other cucurbits. Future functional validation will help to clarify these differences.
Data availability
The bitter gourd genome data have been deposited in the CNGB Nucleotide Sequence Archive (CNSA) (https://db.cngb.org/cnsa/home/; accession: CNP0000016). The RAD data have been deposited at CNGB (https://db.cngb.org/cnsa/; accession: CNP0000012) and the European Nucleotide Archive (ENA) (https://www.ebi.ac.uk/ena/data/view/PRJEB23602).
References
Schaefer, H. & Renner, S. S. A three-genome phylogeny of Momordica (Cucurbitaceae) suggests seven returns from dioecy to monoecy and recent long-distance dispersal to Asia. Mol. Phylogenet. Evol. 54, 553–560 (2010).
Decker-Walters, D. S. Cucurbits, Sanskrit, and the Indo-Aryas. Econ. Bot. 53, 98–112 (1999).
Willdenow, C. L. Linnaei Species Plantarum http://www.botanicus.org/title/b1206998x (1805).
Rheede, H. v. Hortus Malabaricus http://hortusmalabaricus.net/ (1688).
John, K. J. & Antony, V. T. Collection and preliminary evaluation of small bitter gourds (Momordica charantia L.) a relict vegetable of Southern Peninsular India. Genet. Resour. Crop Evol. 56, 99–104 (2009).
John, K. J. & Antony, V. T. A taxonomic revision of the genus Momordica L.(Cucurbitaceae) in India. Indian. J. Plant Genet. Resour. 23, 172–184 (2010).
Chakravarty, H. Fascicles of Flora of India, Fascicle 11:Cucurbitaceae. 87–95 (Botanical Survey of India, Calcutta, 1982).
Marr, K. L., Mei, X. Y. & Bhattarai, N. K. Allozyme, morphological and nutritional analysis bearing on the domestication of Momordica charantia L. (Cucurbitaceae). Econ. Bot. 58, 435–455 (2004).
Behera, T. K. et al. Bitter gourd: botany, horticulture, breeding. Hortic. Rev. 37, 101–141 (2010).
Chen, J. C., Chiu, M. H., Nie, R. L., Cordell, G. A. & Qiu, S. X. Cucurbitacins and cucurbitane glycosides: structures and biological activities. Nat. Prod. Rep. 22, 386–399 (2005).
Tan, M. J. et al. Antidiabetic activities of triterpenoids isolated from bitter melon associated with activation of the AMPK pathway. Chem. Biol. 15, 263–273 (2008).
Amirthaveni, M., Premakumari, S., Gomathi, K. & Yang, R. Hypoglycemic effect of bitter gourd (Momordica charantia L.) among pre diabetics in India: a randomized placebo controlled cross over study. Indian. J. Nutr. Diet. 55, 44–63 (2018).
Krawinkel, M. B. et al. Bitter gourd reduces elevated fasting plasma glucose levels in an intervention study among prediabteics. J. Ethnopharmacol. 216, 1–7 (2018).
Dhillon, N. P. S., Sanguansil, S., Schafleitner, R., Wang, Y. W. & Mccreight, J. D. Diversity among a wide Asian collection of bitter gourd landraces and their genetic relationships with commercial hybrid cultivars. J. Am. Soc. Hort. Sci. 141, 475–484 (2016).
Urasaki, N. et al. Draft genome sequence of bitter gourd (Momordica charantia), a vegetable and medicinal plant in tropical and subtropical regions. DNA Res. 24, 51–58 (2017).
Huang, X. et al. A map of rice genome variation reveals the origin of cultivated rice. Nature 490, 497–501 (2012).
Hufford, M. B. et al. Comparative population genomics of maize domestication and improvement. Nat. Genet. 44, 808–811 (2012).
Zhou, Z. et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 33, 408–414 (2015).
Qi, J. et al. A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nat. Genet. 45, 1510–1515 (2013).
Lin, T. et al. Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 46, 1220–1226 (2014).
Li, R. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272 (2010).
Cui, J. et al. A RAD-based genetic map for anchoring scaffold sequences and identifying QTLs in bitter gourd (Momordica charantia). Front. Plant Sci. 9, 477 (2018).
Li, R. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967 (2009).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 12, 323 (2011).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Smit, A., Hubley, R. & Green, P. RepeatMasker Open-3.0 http://www.repeatmasker.org (1996).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, 265–268 (2007).
Edgar, R. C. & Myers, E. W. PILER: identification and classification of genomic repeats. Bioinformatics 21(Suppl 1), i152–i158 (2005).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1), i351–i358 (2005).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Res. 14, 988–995 (2004).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Elsik, C. G. et al. Creating a honey bee consensus gene set. Genome Biol. 8, R13 (2007).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48 (2000).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27, 29–34 (2000).
Zdobnov, E. M. & Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Li, L., Stoeckert, C. J. Jr. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Wikstrom, N., Savolainen, V. & Chase, M. W. Evolution of the angiosperms: calibrating the family tree. Proc. Biol. Sci. 268, 2211–2220 (2001).
Sun, H. et al. Karyotype stability and unbiased fractionation in the paleo-allotetraploid cucurbita genomes. Mol. Plant 10, 1293–1306 (2017).
Sebastian, P., Schaefer, H., Telford, I. R. & Renner, S. S. Cucumber (Cucumis sativus) and melon (C. melo) have numerous wild relatives in Asia and Australia, and the sister species of melon is from Australia. Proc. Natl Acad. Sci. USA 107, 14269–14273 (2010).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Hasegawa, M., Kishino, H. & Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22, 160–174 (1985).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Mckenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Tang, H., Peng, J., Wang, P. & Risch, N. J. Estimation of individual admixture: analytical and study design considerations. Genet. Epidemiol. 28, 289–301 (2005).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
Schiffels, S. & Durbin, R. Inferring human population size and separation history from multiple genome sequences. Nat. Genet. 46, 919–925 (2014).
Ossowski, S. et al. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327, 92–94 (2010).
Watterson, G. A. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7, 256–276 (1975).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Akey, J. M., Zhang, G., Zhang, K., Jin, L. & Shriver, M. D. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 12, 1805–1814 (2002).
Chen, H., Patterson, N. & Reich, D. Population differentiation as a test for selective sweeps. Genome Res. 20, 393–402 (2010).
Huang, S. et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 41, 1275–1281 (2009).
Garcia-Mas, J. et al. The genome of melon (Cucumis melo L.). Proc. Natl Acad. Sci. USA 109, 11872–11877 (2012).
Guo, S. et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 45, 51–58 (2013).
Monteropau, J. et al. De novo assembly of the zucchini genome reveals a whole‐genome duplication associated with the origin of the Cucurbita genus. Plant Biotechnol. J. 16, 1161–1171 (2018).
Wu, S. et al. The bottle gourd genome provides insights into Cucurbitaceae evolution and facilitates mapping of a Papaya ring-spot virus resistance locus. Plant J. 92, 963–975 (2017).
Martínez-García, P. J. et al. The walnut (Juglans regia) genome sequence reveals diversity in genes coding for the biosynthesis of nonstructural polyphenols. Plant J. 87, 507–532 (2016).
Schaefer, H., Heibl, C. & Renner, S. S. Gourds afloat: a dated phylogeny reveals an Asian origin of the gourd family (Cucurbitaceae) and numerous oversea dispersal events. Proc. R. Soc. B 276, 843–851 (2009).
Cui, J. et al. Genome-wide analysis of simple sequence repeats in bitter gourd (Momordica charantia). Front. Plant Sci. 8, 1103 (2017).
Doebley, J. F., Gaut, B. S. & Smith, B. D. The molecular genetics of crop domestication. Cell 127, 1309–1321 (2006).
Przeworski, M., Hudson, R. R. & Di, R. A. Adjusting the focus on human variation. Trends Genet. 16, 296–302 (2000).
Meyer, R. S., Duval, A. E. & Jensen, H. R. Patterns and processes in crop domestication: an historical review and quantitative analysis of 203 global food crops. N. Phytol. 196, 29–48 (2012).
Phillips, D. R., Rasbery, J. M., Bartel, B. & Matsuda, S. P. Biosynthetic diversity in plant triterpene cyclization. Curr. Opin. Plant Bio. 9, 305–314 (2006).
Shibuya, M., Adachi, S. & Ebizuka, Y. Cucurbitadienol synthase, the first committed enzyme for cucurbitacin biosynthesis, is a distinct enzyme from cycloartenol synthase for phytosterol biosynthesis. Tetrahedron 60, 6995–7003 (2004).
Shang, Y. et al. Biosynthesis, regulation, and domestication of bitterness in cucumber. Science 346, 1084–1088 (2014).
Davidovich-Rikanati, R. et al. Recombinant yeast as a functional tool for understanding bitterness and cucurbitacin biosynthesis in watermelon (Citrullus spp.). Yeast 32, 103–114 (2015).
Takase, S. et al. Identification of triterpene biosynthetic genes from Momordica charantia using RNA-seq analysis. Biosci. Biotech. Bioch. 83, 251–261 (2019).
Zhou, Y. et al. Convergence and divergence of bitterness biosynthesis and regulation in Cucurbitaceae. Nat. Plants 2, 16183 (2016).
Itkin, M. et al. The biosynthetic pathway of the nonsugar, high-intensity sweetener mogroside V from Siraitiagrosvenorii. Proc. Natl Acad. Sci. USA 113, 7619–7628 (2016).
Xie, D. et al. The wax gourd genomes offer insights into the genetic diversity and ancestral cucurbit karyotype. Nat. Commun. 10, 5158 (2019).
Enoch, A. D., Sognigb, N., Adam, A., Jean, G. & Frank, B. Phenetic analysis of wild populations of Momordica charantia L. (Cucurbitaceae) in West Africa and inference of the definition of the new subspecies macroloba Achigan-Dako & Blattner. Candollea 63, 153–167 (2008).
Thimmappa, R., Geisler, K., Louveau, T., O’Maille, P. & Osbourn, A. Triterpene biosynthesis in plants. Annu. Rev. Plant Biol. 65, 225–257 (2014).
Osbourn, A. Secondary metabolic gene clusters: evolutionary toolkits for chemical innovation. Trends Genet. 26, 449–457 (2010).
Kliebenstein, D. J. & Osbourn, A. Making new molecules–evolution of pathways for novel metabolites in plants. Curr. Opin. Plant Biol. 15, 415–423 (2012).
Acknowledgements
We wish to acknowledge the World Vegetable Center (AVRDC), East and Southeast Asia, Thailand, for kindly providing bitter gourd materials for this study. Narinder Dhillon was supported by long-term strategic donors to the World Vegetable Center: the People’s Republic of China (Taiwan), UK aid from the UK government, United States Agency for International Development (USAID), Australian Center for International Agricultural Research (ACIAR), Germany, Thailand, the Philippines, Korea, and Japan. We thank Prof. Dianxiang Zhang of the South China Botanical Garden and Zhengguo Liu of Guangxi University for providing bitter gourd materials. We especially thank Prof. Susanne S. Renner at the University of Munich for providing bitter gourd materials and critical reading of the manuscript. This work was supported by funding from the Key Project of Basic and Applied Research for Ordinary Universities of Guangdong Province (2018KZDXM016), Modern Agricultural Industry Technology System of Guangdong Province, China (2016LM1108, 2017LM1108, and 2018LM1108), Science and Technology Planning Project of Guangdong Province, China (2014B020202006), Central Public-interest Scientific Institution Basal Research Fund for the Chinese Academy of Tropical Agricultural Sciences (No. 1630032015015 and No. 1630032017027), Key Research & Development Program of Guangxi (Guike AB16380059) and Science and Technology Major Project of Guangxi (Guike AA17204026).
Author information
Authors and Affiliations
Contributions
J.Cui and K.H. conceived the project. J.Cui, J.Cheng, W.H., N.P.S.D., and K.H. designed the research. R.H., Q.W., X.H., N.M., J.Cui, J.Cheng, Y.Y., Z.D., D.S., F.H., Z.L., C.Z., C.F., H.Z., J.S., X.W., Y.N. and X.Z. performed the research. J.Cui, Y.Y., S.L., and L.W. wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cui, J., Yang, Y., Luo, S. et al. Whole-genome sequencing provides insights into the genetic diversity and domestication of bitter gourd (Momordica spp.). Hortic Res 7, 85 (2020). https://doi.org/10.1038/s41438-020-0305-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-020-0305-5
