
Abstract
Lithospermum erythrorhizon (red gromwell; zicao) is a medicinal and economically valuable plant belonging to the Boraginaceae family. Roots from L. erythrorhizon have been used for centuries based on the antiviral and wound-healing properties produced from the bioactive compound shikonin and its derivatives. More recently, shikonin, its enantiomer alkannin, and several other shikonin/alkannin derivatives have collectively emerged as valuable natural colorants and as novel drug scaffolds. Despite several transcriptomes and proteomes having been generated from L. erythrorhizon, a reference genome is still unavailable. This has limited investigations into elucidating the shikonin/alkannin pathway and understanding its evolutionary and ecological significance. In this study, we obtained a de novo genome assembly for L. erythrorhizon using a combination of Oxford Nanopore long-read and Illumina short-read sequencing technologies. The resulting genome is ∼367.41 Mb long, with a contig N50 size of 314.31 kb and 27,720 predicted protein-coding genes. Using the L. erythrorhizon genome, we identified several additional p-hydroxybenzoate:geranyltransferase (PGT) homologs and provide insight into their evolutionary history. Phylogenetic analysis of prenyltransferases suggests that PGTs originated in a common ancestor of modern shikonin/alkannin-producing Boraginaceous species, likely from a retrotransposition-derived duplication event of an ancestral prenyltransferase gene. Furthermore, knocking down expression of LePGT1 in L. erythrorhizon hairy root lines revealed that LePGT1 is predominantly responsible for shikonin production early in culture establishment. Taken together, the reference genome reported in this study and the provided analysis on the evolutionary origin of shikonin/alkannin biosynthesis will guide elucidation of the remainder of the pathway.
Similar content being viewed by others

Introduction
The purple-colored roots of red gromwell (Lithospermum erythrorhizon; Fig. 1a, b), also known as “zicao” in Chinese, “jichi” in Korean, and “murasaki” in Japanese, have been used as part of traditional medicines, as a dyestuff, and in cosmetics across many cultures for centuries. The responsible bioactive and pigmented compounds, shikonin—or its enantiomer, alkannin—and dozens of other acylated shikonin/alkannin derivatives (Fig. 1c), are synthesized in the root periderm of L. erythrorhizon and several other Boraginaceae species1,2. Shikonins/alkannins are deposited into the rhizosphere where they function in plant-microbe interactions and interfere with the growth of competing plants (allelopathy), roles suggested to have contributed to the invasion success of species like Echium plantagineum3. The presence of alkannins was also reported in Plagiobothrys arizonicus leaves4, though the physiological and/or ecological significance of their presence in aerial tissues is unclear.

a Intact roots of L. erythrorhizon producing shikonin. b Hairy root culture of L. erythrorhizon producing shikonin. c The structures of shikonin and its enantiomer, alkannin. Shikonin and alkannin are precursor to dozens of acylated derivatives collectively produced by members of the Boraginaceae. d The shikonin pathway starts with the conjugation of 4-hydroxybenzoic acid (4HBA) and geranyl diphosphate (GPP) catalyzed by p-hydroxybenzoate:geranyltransferase (PGT)
Shikonins/alkannins have more recently been discovered to exhibit a range of pharmacological properties5. Shikonin has been found to suppress human immunodeficiency virus (HIV) type 16 and to display anti-tumor effects in breast cancer cells via multiple signaling pathways7. Thus, combined with their traditional medicinal and cosmetic value, there has been wide interest for many decades in scaling shikonin/alkannin production. Early efforts back in the 1970s and 1980s centered on producing shikonin in L. erythrorhizon cell cultures, which was also the first industrial scale platform for producing a secondary metabolite in dedifferentiated plant cells8. With advances in understanding of pathway precursors4,9, strategies to increase shikonin production in L. erythrorhizon through metabolic engineering were developed (e.g.,10,11). Current efforts have extended to include synthetic chemistry for producing shikonin, alkannin, and derivatives with higher specificity and potency12,13. Moreover, the use of comparative transcriptomics (e.g.,14,15,16) and proteomics17 approaches for elucidating the shikonin/alkannin pathway has uncovered several gene candidates and led to the identification of the geranylhydroquinone hydroxylase (GHQH; CYP76B74)18. Comparatively less attention has focused on the evolutionary origin of shikonin/alkannin pathway genes16,19 and on the ecological significance of producing the compounds3. It was only recently that the E. plantagineum genome was published, the first from a shikonin/alkannin-producing species or a member of the Boraginales20.
Despite being among the longest-studied plant natural products, there are still many gaps in knowledge about shikonin/alkannin pathway genes, architecture, and regulation. In this study, we report the first de novo genome for L. erythrorhizon, generated by combining Oxford Nanopore Technology (ONT) long reads with Illumina HiSeq short reads. From this assembly, we identified the existence of 11 previously unreported p-hydroxybenzoate:geranyltransferase (PGT; Fig. 1c) homologs and provide insight into their contribution to shikonin biosynthesis based on (i) their distribution amongst shikonin/alkannin-producing species within the Boraginaceae and (ii) L. erythrorhizon knock-down hairy root lines with reduced expression of LePGT1. Taken together, the results of our study provide evolutionary insight into the origin of the shikonin/alkannin pathway, and the genome assembly offers a major resource for exploring outstanding questions in shikonin/alkannin metabolism.
Results
Genome assembly and annotation
To create a reference genome, we combined L. erythrorhizon ONT genomic DNA (gDNA) reads generated in-house from Siebold & Zucc. plants with publicly available Illumina gDNA reads sequenced by Nanjing University in 2018 from an unknown accession (SRR5644206). The Illumina data consisted of ~21.7 Gb Illumina HiSeq paired-end short reads (150 bp) with an estimated heterozygosity of 0.39% and projected genome size of 369.34 Mb (Fig. S1a). Our in-house ONT data consisted of ~7.6 Gb long-reads (N50 = 15.03 kb) providing roughly 20-fold genome coverage. The short and long reads assembled into 2465 contigs using the DBG2OLC hybrid assembler21, yielding a 367.41 Mb genome with a longest contig of 3.44 Mb and an N50 contig length of 314.31 kb (Table 1).
Using a de novo repeat modeler, 51.78% of assembly bases were denoted as repetitive elements and were subsequently masked (Table S1). Of these elements, the majority were long terminal repeats (LTRs) which comprised 23.43% of the genome assembly. Unclassified elements were the second most common, accounting for 21% of the genome. DNA repeat elements comprised 4.45% of the genome. The repeat content in L. erythrorhizon is comparable to the repeat content observed in the other sequenced Boraginaceae, E. plantagineum (43.3% repetitive; 23.08% LTRs)20.
Protein-coding genes were identified through a combination of ab initio, homology-based, and transcriptome-based prediction methods. A total of 27,720 genes encoding 39,395 proteins were predicted (Table 1). The average protein-coding gene was 3772 bp long and contained 7 exons. Functional annotations were assigned to 80.02%, 72.89%, 59.30%, 23.57%, 7.53%, 5.85% of genes using the InterPro22, Pfam23, GO24, Trans Membrane (TMHMM), KEGG, and MetaCyc25 databases, respectively (Supplementary Dataset S1).
Quality assessment
To evaluate the completeness and coverage of the assembly, we aligned the ONT gDNA, Illumina gDNA, and Illumina RNA-seq reads to the L. erythrorhizon genome assembly. Coverage histograms of the ONT and Illumina gDNA reads showed a single peak at ~18-fold and ~45-fold coverage, respectively (Fig. S1b, c), indicative of the genome being largely homozygous. The alignment rates of the Illumina gDNA reads was high (95.97%); however 46% of the assembled genome lacked gDNA read support (Fig. S1c). This is likely due to the Illumina gDNA reads being generated via a PCR-amplified library, leading to inconsistent coverage across the genome. However, the amount of coding-regions with Illumina gDNA read support was 89.6% (≥10 mapped reads), allowing for sufficient base corrections of the ONT reads in the genic regions, which are the focus of our study. The alignment rate of the RNA reads ranged from 59.29% to 72.71% (in the case of libraries prepared via ribosomal depletion) to 86.74% to 90.15% (for libraries prepared via polyA capture) (Table S2).
We then used BUSCO26 to assess the completeness of the predicted proteome. Within the L. erythrorhizon protein-coding gene set, 1142 of 1400 conserved embryophyta genes (79.3%) were identified as complete, of those 93.43% were present in single-copy and 6.57% were duplicated (Table S3). Furthermore, 279 of 303 conserved eukaryota genes (92.08%) were identified as complete, of those 78.14% were present in single-copy and 21.86% were duplicated (Table S3). Lastly, we calculated the Alien Index (AI) for all predicted proteins of the genome assembly to assess possible contamination27. No assembly contig had a majority of their predicted proteins with AI scores >0, indicating no detectible contamination in the assembly. Only 50 of the 39,395 total proteins (0.13%) had AI scores >0.05, which could be indicative of horizontal gene transfer (HGT) (Supplementary Dataset S2). However, manual inspection of each of these proteins did not yield any strong HGT candidates.
Gene family analysis
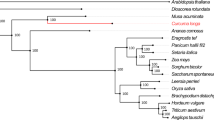
To investigate the evolution of different gene families, including those involved in the production of shikonin, we performed an OrthoFinder28 analysis using the protein-coding genes of L. erythrorhizon and 31 other eudicot species (Table S4). Incorporated into our analysis were four additional Boraginaceae species, including three species known to produce shikonin (E. plantagineum, Arnebia euchroma, and Lithospermum officinale) as well as Mertensia paniculate, a Boraginaceae whose transcriptome was sequenced by the 1000 Plants Initiative (oneKP29) and whose ability to produce shikonin is unknown. We also included 14 additional Boraginales species sequenced by the oneKP project (Table S4). Our OrthoFinder-inferred species tree had the Boraginales sister to a large clade consisting of the Solanales, Gentianales, and Laminales (Fig. 2). This placement is in disagreement with the analysis by Tang et al20. that showed the Boraginales sister to the Solanales. This conflict is unsurprising as the evolutionary relationships of these lamiid orders remains uncertain30. Additional work is needed to resolve these relationships and determine the source of the phylogenetic discordance.

The OrthoFinder inferred species phylogeny is displayed on the left. The branch thickness is scaled based on the number of predicted duplicated events to have occurred at the descendent node; thinner branches indicate fewer duplications, thicker branches indicate more (see Table S7). Internodes discussed in the text (N1, N5, N10, and N15) are labeled (the species tree with all labeled internodes can be accessed in Table S7). Asterisks (*) indicate known shikonin/alkannin-producing species. Horizontal bar plots (right) indicate the number of orthogroups that are species-specific (red), maintained in all 32 species (green), or present in more than one but less than all species in the analysis (blue)
The OrthoFinder analysis identified 100,874 orthogroups (predicted gene families), of which 24,346 consisted of two or more species in the analysis (Table S5). Of the 14,885 orthogroups containing one or more L. erythrorhizon sequences, 3441 orthogroups (23.13%) were present in all species, 3003 (20.17%) were L. erythrorhizon specific, and 8441 (56.71%) consisted of L. erythrorhizon and one or more additional species (Fig. 2; Table S6). In total, 36,392 of 39,395 L. erythrorhizon proteins (92.38%) were assigned to an orthogroup containing sequence(s) from one or more additional species (Table S6; Supplementary Dataset S3). The total number of L. erythrorhizon orthogroups (14,885) was comparable to the other sequenced genomes in the analysis (average 15,227), which ranged from 12,666 in Solanum lycopersicum (tomato) to 19,349 in Actinidia chinensis (kiwi). Furthermore, the percentage of L. erythrorhizon orthogroups that was species-specific (20.17%) was also comparable to the other sequenced genomes (average 22.47%), ranging from 5.05% in tomato to 46.55% in Utricularia gibba (a carnivorous aquatic bladderwort). The other three species known to produce shikonin showed an increased number of orthogroups (average 26,040.33), which is likely a result of their predicted proteomes being derived from de novo transcriptome assemblies rather than sequenced genomes. The average percentage of species-specific orthogroups found in the oneKP transcriptomes (0.50%) was noticeably lower that the other species in the analysis (Fig. 2) due to these proteomes being filtered prior to publication29.
To identify orthogroups that had expanded in one or more ancestors of L. erythrorhizon, we parsed the number of OrthoFinder-predicted gene duplications at each node of the inferred species tree (Fig. 2; Table S7). The average number of orthogroups that duplicated one or more times at a given internode (i.e., non-leaf node) was 1,489.55 and ranged from 41 duplications at internode N5 (the common ancestor of Daucus carota and A. chinensis) to 4489 duplications at internode N1 (the common ancestor of Caryophyllales and asterids) (Table S7). A total of 2818 orthogroups duplicated at internode N10 (the common ancestor of the five Boraginaceae species), and the L. erythrorhizon genes that duplicated at this internode were enriched in 21 gene ontology (GO) categories (Benjamini–Hochberg adjusted p-value < 0.1; Table S8) including transferase activity (GO:0016740; p = 1.10e−3), signal transduction (GO:0007165; p = 6.45e−4), and transmembrane transporter activity (GO:0022857; p = 0.08). Similarly, the L. erythrorhizon genes from the 3908 orthogroups that duplicated at internode N15 (the common ancestor of the four Boraginaceae species known to produce shikonin) were enriched in 14 GO categories (Benjamini–Hochberg adjusted p-value < 0.1; Table S8) including transferase activity of acyl groups (GO:0016746; p = 0.3), DNA-binding transcription factor activity (GO:0003700; p = 3.79e−3), and transmembrane transport (GO:0055085; p = 0.07).
Evolution of p-hydroxybenzoate:geranyltransferase (PGT) genes for shikonin biosynthesis
One orthogroup predicted to have undergone multiple duplication events in the last common ancestor of shikonin/alkannin-producing species was OG0000509. This orthogroup was comprised of genes that code for prenyltransferases, including the characterized ubiquinone prenyltransferase in Arabidopsis thaliana, AtPPT1 (Coq2; At4g23660)31. In addition, OG0000509 contained 13 L. erythrorhizon genes (Table S9), including Leryth_015068 (hereafter referred to as LePGT1), which encodes a protein 97.06% identical to LePGT1, and Leryth_002561 (hereafter referred to as LePGT2), which encodes a protein 100% identical to LePGT232. The p-hydroxybenzoate:geranyltransferases (PGTs) catalyze the conjugation of 4-hydroxybenzoic acid (4HBA) and geranyl diphosphate (GPP), the first and committed step of the shikonin pathway (Fig. 1d)32,33,34 and have been identified from several shikonin/alkannin-producing species19,35,36,37.
To better understand the evolutionary history of LePGT1 and LePGT2, we constructed a robust phylogenetic tree of OG0000509 prenyltransferases (Fig. 3a). Of the 13 sequences in L. erythrorhizon, one was excluded from the phylogenetic analysis due to it being a suspected pseudogene (Leryth_015069), which is located ~10 kb away from LePGT1 and is likely the result of a tandem duplication event (Fig. S2). Pseudogenization is suspected due to the fact that the sequence appears to be truncated and is missing the conserved NDXXD motif indicative of putative prenyl diphosphate binding (Fig. S3)32. In agreement with the OrthoFinder analysis, the phylogeny shows a large radiation of prenyltransferase genes in the Boraginales followed by an additional radiation in the Boraginaceae. The Leryth_011786 gene copy is notable in that it is on a small branch relative to the other L. erythrorhizon sequences and groups closest to homologs in the other lamiids (Fig. 3a), suggesting it is likely the “missing” ubiquinone prenyltransferase38 (hereafter referred to LePPT1).

a Maximum likelihood tree of orthogroup OG0000509 rooted on Arabidopsis thaliana ubiquinone polyprenyltransferase, AtPPT1. Nodes with IQ-TREE ultrafast parametric support values >0.95 are indicated by gray circles on the preceding branch. The branches and outer color bar are color-coded to match the taxonomic classification of each sequence. Lithospermum erythrorhizon sequences are indicated by the red font. The hypothetical location of the inferred duplication via retrotransposition in the ancestor of genes encoding PGTs and PGT-like homologs is indicated by the gray arrow. b Heatmap showing the gene expression pattern of L. erythrorhizon prenyltransferase genes in whole roots (R), aerial tissue (A), root periderm (P), root vascular (V), hairy root grown in the dark (D), and hairy root grown in the light (L). Conditions where shikonin is most abundant are indicated with an aterisk (*). The cladogram (left) shows the evolutionary relationship between prenyltransferase genes according to the overall maximum likelihood phylogeny in part a
As recognized by Kusano et al.38, LePGT1 and LePGT2 are both comprised of a single exon (Fig. S2). The six L. erythrorhizon genes most closely related to LePGT1 and LePGT2 (Leryth_008226, Leryth_027669, Leryth_027448, Leryth_000057, Leryth_020476, Leryth_003298) are also single exonic. In contrast, AtPPT1, as well as its predicted L. erythrorhizon ortholog, LePPT1, are multi-exonic, each containing eight exons. The three remaining unclassified L. erythrorhizon genes in the phylogeny were found to contain a variable number of exons: Leryth_023194 with ten exons, Leryth_011510 with eight exons, and Leryth_006101 with ten exons. The loss of exons in LePGT1 and LePGT2, and the six additional PGT-like genes, suggests that a retrotranspotition event is responsible for the duplication that gave rise to the specialized prenyltransferase genes involved in the production of shikonin.
In order to investigate the mechanisms and timing of the PGT gene family expansion, we identified syntenic blocks for the entire L. erythrorhizon genome assembly and performed synonymous substitution (Ks) pattern analyses. Regions of shared Ks values among homologous genes within syntenic blocks would be indicative of a possible whole-genome duplication (WGD) in the ancestor of L. erythrorhizon. We identified a (Ks) peak at roughly ~0.45, a value that is suggestive of a potential polyploidy event39 (Fig. S4a), and roughly matches the results of Tang et al., who performed a larger analysis of WGD in the Boraginaceae. In the Tang analysis, the Ks peak of ~0.417 was proposed to have arisen via a WGD in the ancestor of the Boraginaceae roughly 25 MYA20. Given evidence of a probable WGD in the ancestor of L. erythrorrhizon, the expansion of the OG0000509 genes could have arisen due to the WGD or subsequent duplication processes (e.g., tandem duplication, retrotransposition). However, only two syntenic blocks containing PGT and PGT-like genes were identified (Table S10). The first syntenic block had a median Ks of 0.534 and contained PGT1 (Leryth_015168) and another single exonic PGT-like gene (Leryth_008226) (Fig. S4b). The second syntenic block had a median Ks of 0.466 and contained two multi-exonic PGT-like genes (Leryth_011510 and Leryth_006101) (Fig. S4c). Given that the median Ks of the two syntenic blocks containing PGT and PGT-like genes are approximate to the multi-locus peak (~0.45) in the Ks distribution (Fig. S4a), it is possible that these duplications arose via the WGD event. Though, the fact that there is shared synteny between two multi-exonic homologs as well as between two single-exonic homologs, while no evidence of shared synteny between multi-exonic homologs and single-exonic homologs was observed, suggests that the putative retrotransposition event occurred prior to the whole-genome duplication. This apparent lack of synteny also indicates that the contigs harboring PGT homologs are not simply haplotypes resulting from the genome assembly process, but that these genes are found on contigs with unique neighboring genetic sequence.
Aside from the suspected pseudogene, all other encoded prenyltransferases in orthogroup OG0000509 contain the conserved NDXXD motif indicative of putative prenyl diphosphate binding (Fig. S3)32. All but two unclassified homologs (Leryth_023194 and Leryth_006101) also maintained the GX(K/Y)STAL sequence motif conserved in this subfamily of 4HB:prenyltransferases (Fig. S3)32. One of these unclassified homologs, Leryth_023194, also contained an N-terminal chloroplast targeting sequence (Table S11). None of the other prenyltransferase homologs contained a detectable signal or transit peptide. Lastly, we checked the relative gene expression of all prenyltransferase homologs in three tissue/growth condition comparisons in which shikonin levels and/or expression of shikonin pathway genes are variably abundant: L. erythrorhizon whole root tissue versus aboveground tissue37, root outer periderm versus inner vascular tissue (Fig. S5), and hairy root tissue cultures grown in M9 in the dark versus in B5 in the light40. LePGT1 and LePGT2, along with PGT-like homologs Leryth_027448 and Leryth_027669 were significantly overrepresented (adjusted p value < 0.05, log2-fold change >1) in conditions associated with known increased shikonin production or shikonin pathway gene expression states (whole root tissue, root periderm tissue, and hairy root grown in M9 in the dark; Fig. 3b; Table S9). Additionally, PGT-like homolog Leryth_020476 was significantly overrepresented in whole root and root periderm tissue (Fig. 3b; Table S9). Lastly, Leryth_000057 and Leyrth_023194 were significantly overrepresented in root periderm tissue. The remaining PGT homologs showed zero to low expression in all samples.
LePGT1 is the predominant PGT functioning in the shikonin pathway
LePGT1 is considered as the key regulatory enzyme in the shikonin pathway41,42. As there are no reported genetic studies with PGTs, and in light of the newly identified PGT-like encoding genes found in the L. erythrorhizon genome (Fig. 4), we knocked down LePGT1 expression in L. erythrorhizon hairy roots to investigate if LePGT1 is indeed the predominant PGT controlling shikonin production. Several independent PGT1-RNAi (PGT1i) lines were generated, excised, transferred to B5 media plates for selection, and then screened based on total shikonins (the sum of shikonin plus its derivatives) production in liquid culture using HPLC coupled with diode array detection (DAD). Individual lines were cultured in liquid B5 in constant light without selection for 14 d and then transferred to M9 and constant darkness to induce shikonin production. Analysis of culture media 3 d after transfer to M9 and darkness revealed 17 PGT1i lines producing between 1 and 59% of the total shikonins synthesized by control hairy roots sampled at the same time (Fig. 4a). Two lines, PGT1i-21 and PGT1i-79, were further analyzed by qRT-PCR. Both lines were found to have greater than 95% reduced LePGT1 levels while expression of LePGT2 remained statistically unchanged compared to the control (Fig. 4b). Because the gene encoding LePGT1 is more similar to LePGT2 than any of the PGT-likes (Fig. 3a), these data indicate that the RNAi construct specifically targeted LePGT1. Analysis of the culture media from the same hairy roots used to perform qRT-PCR revealed that the reduction in LePGT1 expression (Fig. 4b) correlated with a more than 95% decrease in total shikonin content (Fig. 4c). These results provide further support for LePGT1 being predominantly responsible for the formation of 3-geranyl-4HBA. They also imply that the PGT-like proteins encoded in the L. erythrorhizon genome likely do not play a major role in shikonin production.

a Screening of LePGT1-RNAi (PGT1i) lines based on total shikonin levels present in liquid culture media 3 d after transfer of 14-d-old hairy roots to M9 and darkness. b Expression levels of LePGT1 and LePGT2 in hairy roots of two independent PGT1i lines compared to control. c Analysis of total shikonin in same lines used to measure expression in panel b. All data are means ± SEM (n = 3–4 biological replicates). Different letters indicate significant differences via analysis of variance (ANOVA) followed by post-hoc Tukey test (α = 0.05). In b, lowercase and capital letters correspond to statistical comparisons for LePGT1 and LePGT2 expression, respectively
Discussion
In this study, we report the first de novo assembly of a L. erythrorhizon genome obtained through a combination of ONT long-read and Illumina HiSeq short-read sequencing technologies. The ~367.41 Mb assembly contained 27,720 predicted protein-coding genes that were clustered into 14,885 orthologous gene families, 79.84% of which were also found in other species in our comparative phylogenetic analysis (Fig. 2). A significant number of these orthogroups (2818) appear to have duplicated in the last common ancestor of the five Boraginaceae species in our analysis, and another 3908 orthogroups appear to have duplicated in the last common ancestor of the four known shikonin/alkannin-producing species. Among the 6726 orthogroups duplicated at these internodes is OG0000509, the orthogroup containing AtPPT1 and LePPT1 (genes involved in the primary metabolic process of ubiquinone biosynthesis) as well as the specialized genes LePGT1 and LePGT2 involved in shikonin biosynthesis. An additional nine homologous prenyltransferases of unknown function were also identified in the L. erythrorhizon genome. This analysis illustrates the importance of gene duplication in the evolution of the shikonin/alkannin pathway, in agreement with other analyses of specialized metabolism in plants (e.g.,43,44,45). In the case of the PGT and PGT-like sequences in L. erythrorhizon, the initial duplication event appears to have occurred via retrotransposition based on the fact that these sequences appear to have lost all introns (Fig. 3a; Table S9). The relative contribution of retrotransposition compared to other processes that generate gene duplicates (e.g., whole-genome duplication) to metabolic innovation in plants remains to be investigated. Overall, this genomic resource will complement the available E. plantagineum genome20 and the extensive sets of transcriptomes (e.g., refs 14,15,16) and proteomes17 published from Boraginaceae species for elucidating the shikonin/alkannin pathway and its evolutionary origin, as well as the evolution of specialized metabolism more generally in this family.
The connection between shikonin/alkannin and ubiquinone biosynthesis is not limited to homologous prenyltransferases. The hydroxybenzene ring, ring A (Fig. 5), of shikonin/alkannin’s naphthazarin moiety is derived from L-phenylalanine via cinnamic acid and 4HBA4,46–the same route that is partially responsible for forming the benzoquinone ring of ubiquinone (coenzyme Q) in plants47,48. Like the shikonin/alkannin pathway, ubiquinone biosynthesis starts off with the conjugation of 4HBA with a polyprenyldiphosphate, catalyzed by a PPT (Fig. 5). In addition to the analogous PPT- and PGT-catalyzed reactions, the ubiquinone and shikonin/alkannin pathways share other similar ring modification reactions that occur early in their respective pathways (Fig. 5). Both ubiquinone and shikonin/alkannin biosynthesis require the prenylated 4HBA ring to be decarboxylated and hydroxylated at the C1 position, though the sequence of reactions and necessary enzymes are unknown in both pathways. In the Escherichia coli ubiquinone pathway, the non-oxidative ring decarboxylation of 3-polyprenyl-4HBA is catalyzed by UbiD in concert with a UbiX chaperone for substrate reorientation49. Orthologs of genes encoding UbiD and UbiX are absent from plant genomes. Therefore, if plants also use a series of non-oxidative decarboxylation and hydroxylation steps to modify the C1 position of the 4HBA ring, they do so using other evolved enzymes. It is also possible that plants have evolved to achieve the decarboxylation and hydroxylation via an oxidative decarboxylase that would carry out both reactions. Whatever the mechanism for C1 decarboxylation and hydroxylation, if it is indeed shared between shikonin/alkannin and ubiquinone biosynthesis, it is possible that there is an evolutionary linkage among the enzymes involved given the other metabolic connections between the two pathways.

The committed steps of the shikonin and ubiquinone pathways rely on homologous prenyltransferases that conjugate 4-hydroxybenzoic acid (4HBA), derived from phenylalanine, with a prenyl diphosphate precursor. Subsequent decarboxylation and hydroxylation at the C1 position of the 3-prenylated/geranylated 4HBA ring is required in both pathways, although the responsible enzymes (depicted by “?”) in each route remain unknown. Non-oxidative decarboxylation of the prenylated-4HBA ring is shown as it occurs in bacteria. It is possible that plants use an oxidative decarboxylation mechanism, which would result in concomitant decarboxylation and hydroxylation at the C1 position, bypassing the phenolic intermediates. Abbreviations: GHQH, geranylhydroquinone hydroxylase; PGT, p-hydroxybenzoate:geranyltransferase; PPT, polyprenyltransferase
Downregulation of LePGT1 expression by 95% was sufficient to reduce shikonin production by more than 97% 3 d after transfer of hairy roots to M9 media and darkness (Fig. 4b, c). While this provides strong evidence that LePGT1 is principally controlling shikonin formation, it does not rule out that LePGT2 still plays a significant role. Like LePGT1, LePGT2 is highly expressed under shikonin producing conditions (Fig. 3b; Table S9)32,50. One explanation for achieving near abolishment of shikonin by only knocking down LePGT1 could be that LePGT1 and LePGT2 form heteromers in vivo. Biochemical studies, however, suggest that LePGT1 is capable of functioning homomericly41,42, so this appears unlikely. Another possibility is that LePGT1 is the predominant form functioning early in shikonin-producing conditions because of its biochemical properties. LePGT1 has 5- and 10-fold higher affinities for 4HBA and GPP, respectively, compared to LePGT232. It can be envisioned that early in hairy root culture establishment, when precursor concentrations are a priori low, that LePGT1 is the de facto PGT responsible for forming 3-geranyl-4HBA. In this study, shikonin levels were measured at day 3, the first day that visible production occurred in control cultures. It is possible that over time, as precursor pools increase, the relative contribution of LePGT2 would become greater. If any of the PGT-like proteins encoded in the L. erythrorhizon genome also contribute PGT activity, their relative contributions would also increase over the culture period. In light of the current study, reports of multiple PGT unigenes and their variable expression patterns in other transcriptomic studies19,37 should be re-investigated to determine if any encode PGT-like proteins. More detailed investigations looking at shikonin formation over time using metabolic flux analysis with stable isotopic labeling are needed to determine the temporal contributions of PGTs.
Conclusions
The first genome assembly from the medicinally and economically important plant L. erythrorhizon is expected to advance understanding of the evolutionary history of the Boraginales. As just the second genome from a member of this order, the other from E. plantagineum20, the L. erythrorhizon genome will provide another piece of the puzzle needed to reconstruct the phylogenetic relationships among the Boraginales, Solanales, Gentianales, and Laminales (Fig. 2). The L. erythrorhizon genome will also serve as a novel tool for elucidating the remaining missing steps in the shikonin/alkannin pathway and for filling gaps in knowledge about its metabolic origin. Our phylogenetic analysis of prenyltransferases encoded in the L. erythrorhizon genome (Fig. 3a) has already led to the remarkable discovery of several additional encoded PGTs. It also provided evidence to suggest that PGTs arose in a common ancestor of modern shikonin/alkannin-producing Boraginaceae species via a retrotransposition-derived duplication event and subsequent neofunctionalization of an ancestral prenyltransferase gene. Based on homology between PGTs and PPTs it is possible that this points to an evolutionary link between ubiquinone and shikonin/alkannin biosynthesis, especially considering the other metabolic similarities shared between the two pathways (Fig. 5). This would not be the first connection found between primary and specialized quinone metabolism in plants as it was recently reported that the pathway to synthesize the naphthoquinone moiety of juglone in black walnut trees (Juglans nigra) is shared with the phylloquinone (vitamin K1) pathway51. Taken together, the results from our study provide several new leads for investigating the evolution of specialized metabolism in the Boraginaceae.
Materials and methods
Plant materials, growth conditions, and general experimental procedures
Seeds of L. erythrorhizon (accession Siebold & Zucc.) were obtained from the seed bank at the Leibniz Institute of Plant Genetics and Crop Plant Research (IPK) in Germany and plants were propagated under standard greenhouse conditions to bulk seeds.
For all hairy root culture work, L. erythrorhizon Siebold & Zucc. seeds were sterilized by chlorine gas according to Lindsey et al.52. After exposure to chlorine gas for 2 h, seeds were rinsed five times with sterile water and shaken in 200 µg/mL cefotaxime overnight. They were then rinsed with sterile water and plated on half-strength Murashige and Skoog (MS, Phytotech Labs) media with 0.1% Plant Preservation Mixture (PPMTM, Plant Cell Technology). Seeds were stratified at 4 °C for 2 weeks and moved to room temperature under 12:12 light:dark cycle. Once germinated, seed coats were removed, and each seedling was transferred to a magenta box with half-strength MS media. Plants with their second pairs of true leaves were used in hairy root transformation.
Hairy roots were maintained in solid Gamborg B5 media (Phytotech Labs) containing 3% sucrose and 10 mg/L Basta (PlantMediaTM). Hairy roots were subcultured every two weeks. For quantification of shikonin, 1-cm hairy root fragments were transferred to 20 mL liquid Gamborg B5 media and grown under constant light at 28 °C for two weeks without Basta. Shikonin production was then induced by transferring hairy roots to M9 media53 and culturing at 28 °C in constant darkness without Basta.
Nanopore sequencing
For nanopore sequencing, leaves from an in vitro cultured 3-month-old L. erythrorhizon Siebold & Zucc. plant were frozen in liquid nitrogen, ground by mortar and pestle, and high molecular weight genomic DNA was extracted using a CTAB phenol chloroform extraction protocol (available on protocols.io: https://doi.org/10.17504/protocols.io.bamnic5e) and purified using a Genomic DNA Clean and Concentrator kit (Zymo Research). At least 2 μg of gDNA was used as input for an Oxford Nanopore LSK-109 library ligation kit and sequenced on R9 MinION flow cells. Base calling was performed with Guppy v2.3.554. Reads less than 3 kilobase pairs long or with quality scores less than 7 were discarded. Reads are available for download at the NCBI Sequence Read Archive (PRJNA596998).
Genome assembly
Lithospermum erythrorhizon Illumina gDNA PE reads sequenced by Nanjing University in 2018 from an unknown accession were downloaded from NCBI SRA database experiment SRR5644206 and assembled with Abyss v2.1.555 using a k-mer size of 75. The assembled Abyss contigs and the ONT long reads served as input for the DBG2OLC hybrid assembler using the following parameters: KmerCovTh 2, AdaptiveTh 0.0001, MinOverlap 20, RemoveChimera 1, k 1721. The resulting hybrid assembly was error corrected via five rounds of polishing with the Illumina gDNA reads using Pilon v1.2356. Five additional rounds of Pilon polishing were performed using L. erythrorhizon Siebold & Zucc. stranded RNA-seq reads (see RNA-seq experiments section below) as input to fix single nucleotide errors in transcribed regions. Genome size was estimated using GenomeScope57 with a k-mer (k = 21) depth distribution of the Illumina gDNA reads calculated using Jellyfish v2.2.1058. Illumina gDNA reads were mapped back to the final assembly using BWA v0.7.1559, RNAseq reads were mapped using STAR v2.5.460, and ONT reads were mapped using minimap2 v2.1361. Histograms of Illumina and ONT read depth when mapped to the final assembly were generated by the program purge_haplotigs v1.1.1 to assess the level of assembly heterozygosity62.
Genome annotation
De novo repeat identification was performed using RepeatModeler v1.0.9 and masked using RepeatMasker v4.0.7 (http://repeatmasker.org). Gene model and protein prediction was conducted with MAKER2 v2.31.1063 by supplying protein homology-based evidence, transcriptomic evidence in the form of a genome guided transcriptome assembly generated from in-house Siebold & Zucc. stranded RNA-seq reads (see RNA-seq experiments section below) using Trinity v2.5.164, and ab initio gene calling using SNAP65 and BRAKER266. Gene models with an AED score <0.9 or ones that encoded a predicted protein <30 amino acids long were excluded from the final gene set. Additional information on the full annotation pipeline is presented in Fig. S6. Conservation of core genes was performed using BUSCO v2.026. Functional annotation of the final protein set was performed using InterProScan22 and TargetP67.
We checked the genome for possible contamination using the Alien Index (AI) pipeline (https://github.rcac.purdue.edu/jwisecav/phylo-pipe; last updated August 26, 2019) as previously described27. Briefly, each predicted protein sequence was queried against the NCBI RefSeq database (release 97) using Diamond v0.9.22.12368, and the AI score was calculated based on the output. The AI score is given by the formula: AI = nbsO-nbsE, where nbsO is the normalized bit score of the best hit to a species outside of the eudicot lineage, nbsE is the normalized bit score of the best hit to a species within the eudicot lineage (skipping all hits to L. erythrorhizon present in the RefSeq database). AI scores range from -1 to 1, being greater than zero if the predicted protein sequence had a better hit to a non-eudicot species, suggestive of either HGT or contamination27.
RNA-seq experiments
For the L. erythrorhizon root periderm and vascular tissues RNA-seq experiment, 3-month-old Siebold & Zucc. plants grown in soil under standard greenhouse conditions were harvested. Roots were collected from nine individual plants and divided into three groups, each containing three unique individuals. The periderm and vascular tissues were isolated by peeling the periderm from the roots (Fig. S5a), and the prepared portions from the three individuals in each group were pooled. Tissues were frozen in liquid nitrogen, ground by mortar and pestle, and 100 mg was used to analyze total shikonin content each sample (Fig. S5b). From the same sets of samples, RNA was extracted as described below, quantified, and DNase-treated (NEB) according to the manufacturer’s instructions. A total of six cDNA libraries from the three biological replicates prepared from each of the L. erythrorhizon periderm and vascular tissue pools, were constructed using a ribominus TruSeq Stranded Total RNA library prep kit (Illumina, San Diego, CA), and 101-bp paired-end reads were generated via Illumina HiSeq 2500 at the Purdue Genomics Center, with at least 67 million reads per library. Sequence quality was assessed by FastQC (v. 0.10.0; http://www.bioinformatics.babraham.ac.uk). The raw data were submitted to the Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra/) and are available at the NCBI Sequence Read Archive (PRJNA596998).
The experimental design for the RNA-seq experiment comparing L. erythrorhizon hairy roots sampled in B5 in the light and M9 in the dark was based on a previous report of observed rapid increases in expression of shikonin precursor pathway genes, and in PGT, within 2 h after switching L. erythrorhizon cell cultures from growth in B5 in the light to growth in M9 in darkness40. In this study, several cultures from three independently generated L. erythrorhizon hairy root lines were started in liquid Gamborg B5 media containing 3% sucrose at 28 °C in the light (~100 µE m−2 s−1). After 2 weeks, hairy roots from three cultures for each of the three lines (n = 3 biological replicates per line) were harvested and pooled to represent the B5 light-treated samples. The remaining hairy root cultures were transferred to M9 media and darkness. After 2 h, hairy roots from three cultures for each of the three lines (n = 3 biological replicates per line) were harvested and pooled to represent the M9 dark-treated samples. Samples were frozen in liquid nitrogen, ground by mortar and pestle, and RNA was extracted as described below. Six cDNA libraries were generated with a TruSeq Stranded mRNA library prep kit (Illumina, San Diego, CA) and were sequenced on an Illumina NovaSeq 6000 at the Purdue Genomics Center. Sequence quality assessment were performed as described above for the periderm and vascular tissues RNA-seq experiment. The raw data were submitted to the Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra/) and are available at the NCBI Sequence Read Archive (PRJNA596998).
Additionally, unstranded RNA-seq data of L. erythrorhizon whole roots and aerial tissue from an unknown accession was downloaded from the NCBI SRA (experiments SRR3957230 and SRR3957231) to include in the gene expression analysis. Gene abundance estimates of PGT and PGT-like genes (Fig. 3b, Table S9) were measured using Kallisto v0.45.069 and normalized for library depth using DESeq270. Differential expression status was determined using the EdgeR v3.24.371 package. For the EdgeR analysis, raw counts were normalized into effective library sizes using the trimmed mean of M-values (TMM) method72, and exact tests were conducted using a trended dispersion value and a double tail reject region. A false discovery rate was calculated using the Benjamini–Hochberg procedure73. Genes with a log2-fold change in abundance greater than 1 and false discovery rate less than 0.05 were considered as differentially represented.
De novo transcriptome assemblies of additional Boraginaceae species
Illumina RNA-seq reads from additional shikonin-producing species L. officinale, Arnebia euchroma and Echium plantagineum19,37 were downloaded from the following NCBI SRA experiments: SRR4034889, SRR4034892, SRR4034890, SRR4034891, SRR6799516, SRR6799517, and SRR6799518. Raw RNA-seq reads were normalized and error corrected with BBnorm using a target size of 40 and a minimum depth of 2 (software last modified October 19, 2017) and Tadpole using default parameters (software last modified June 27, 2017), programs from the BBMap software package74. The resulting clean reads were assembled de novo using Trinity v2.5.164 with default parameters for stranded (L. officinale) and unstranded (E. plantagineum and A. euchroma) libraries. Coding regions were inferred using TransDecoder v3.0.175.
Identification of orthologous gene families
Homology between the predicted proteomes of L. erythrorhizon and 31 other eudicot species was determined with OrthoFinder v2.1.2 using the following parameters: -S diamond -M msa -T fasttree28. The species tree in Fig. 2 was generated using TreeGraph2 v2.15.076. Hypergeometric tests were performed in python using the SciPy library hypergeom, and p-values were adjusted for multiple comparisons using the StatsModels library multitest with the Benjamini & Hochberg (BH) method73.
Phylogenetic analysis
We performed a separate phylogenetic analysis of the orthogroup containing LePGT1 and LePGT2 (OG0000509). Three sequences were excluded due to their long branches, including the suspected pseudogene Leryth_015069. The remaining sequences were aligned with MAFFT v7.407 using the E-INS-I strategy and following parameters: --maxiterate 1000 --bl 45 --op 1.0 --retree 377. The maximum likelihood phylogeny was constructed using IQ-TREE version 1.6.1078 using the built in ModelFinder to determine the best-fit substitution model79 and performing SH-aLRT and ultrafast bootstrapping analyses with 1,000 replicates each. The gene tree in Fig. 3a was generated using iTOL v480.
Synteny analysis
Regions of shared synteny within the genome of L. erythrorhizon were detected using SynMap2 on the online Comparative Genomics Platform (CoGe) using default settings with the exception that the merge syntenic blocks algorithm was set to Quota Align Merge, the syntenic depth algorithm was set to Quota Align, and the CodeML option was activated to calculate substitution rates between syntenic CDS pairs. For syntenic blocks containing PGT genes and their homologs, the encompasing contigs were aligned using promer (v3.07) of the MUMmer4 alignment system81.
Cloning and generation of LePGT1i hairy root lines
For the LePGT1-RNAi construct, DNA containing two spliced LePGT1 cDNA fragments of the coding region corresponding to nucleotides 179-698 and 179-502, the latter in antisense orientation to create a hairpin structure, was synthesized (Genscript, Piscataway, NJ). 5’-CACC was added for subcloning into pENTRTM/D-TOPO (InvitrogenTM, Carlsbad, CA), sequence verified, and transferred into the destination vector, pB2GW782, by recombination using LR Clonase Enzyme MixTM (Invitrogen). The final construct, pB2GW7-PGT1i, was transformed into Agrobacterium rhizogenes strain ATCC 15834 competent cells by freeze-thaw transformation83. Briefly, competent cells were incubated with 100 ng of pB2GW7-PGT1i for 15 min on ice. Then, cells were snap frozen in liquid nitrogen for 5 min and consecutively thawed at 37 °C for 5 min. Nutrient Broth (NB) media was added to the culture and kept in 37 °C with shaking for 2 h before being plated on NB agar containing 50 µg/mL spectinomycin for selection.
L. erythrorhizon hairy root PGT1i lines were generated based on the protocol from Fang et al.84 with slight modification. A. rhizogenes containing pB2GW7-PGT1i was inoculated in NB with 50 µg/mL spectinomycin and kept in shaking incubator at 28 °C until reached OD600 = 1. Then, acetosyringone was added to the media to a final concentration of 0.1 mM and the culture was grown further for 4 h in dark. The A. rhizogenes was then pelleted by centrifugation. The pellet was washed and resuspended in half-strength MS containing 0.1 mM acetosyringone. Each stem of sterile plants grown in tissue culture was wounded by surgical blade and the prepared culture was applied to the wounded area by cotton swab. The plants were then kept in darkness for 1 d and returned to normal growth conditions. The hairy roots emerged between 10 and 28 d post infection. Emergent roots were excised and placed on Gamborg B5 media with 3% sucrose and 200 µg/mL cefotaxime to eliminate A. rhizogenes. After 2 weeks, hairy roots were transferred to Gamborg B5 media containing 3% sucrose and 10 mg/L Basta for selection for 2 weeks. Hairy root lines transformed by A. rhizogenes without pB2GW7-PGT1i were generated to use as control.
RNA extraction and qRT-PCR
Total RNA was extracted from ~20 mg of hairy root tissues according to the protocol from Ghawana et al.85 Briefly, the samples were frozen by liquid nitrogen and ground by mortar and pestle. 2 mL of RNA extraction buffer (phenol containing 0.1% sodium dodecyl sulfate, 0.32 M sodium acetate, and 0.1 M ethylenediaminetetra acetic acid) was added to the sample in the mortar and mixed, followed by addition 0.8 mL of RNAse-free water. After mixing, the mixture was incubated for 5 min before transfer to microtubes. 0.3 mL of chloroform was added, and the sample was vortexed and centrifuged at 4 °C, 13,000 rpm for 10 min. The supernatant was then transferred to the new tube containing 0.6 mL isopropanol. Next, the sample was mixed by inverting, and nucleic acids were precipitated at −20 °C for 10 min. After precipitation, the sample was centrifuged at 4 °C for 10 min. The pellet was washed with 70% ethanol before air drying. The RNA pellet was dissolved in RNase-free water. The total RNA was concentrated and purified using an RNA Clean & Concentrator Kit (Zymo Research) with on-column DNase treatment (Zymo Research) using the manufacturer protocol. cDNA synthesis was performed by 5X All-In-One RT MasterMix (abm) according to the manufacturer instructions using 500 ng of total RNA.
Expression of LePGT1 and LePGT2 was measured by qRT-PCR with comparative quantification using the 2−ΔΔCT method86. Primers were designed using Primer-BLAST on NCBI87. Due to the sequence similarity of members of the LePGT and LePGT-like (Table S9) gene family, each primer was checked against all members for possible off-target matches. To minimize off-target amplification, primer pairs were selected that had either (i) four or more mismatches in a primer to all other LePGT and LePGT-like family genes or (ii) two mismatches in one primer and three mismatches in the other primer to all other LePGT and LePGT-like family genes88. qRT-PCR reactions were performed using a QuantStudioTM 6 (ThermoFisher) in a 10 μL reaction as follows: 5 μL of 5x Fast SYBR Green PCR master mix (ThermoFisher), 1 μL each of the forward and reverse primers (50–900 nM final concentration; Table S12), and 3 μL of diluted cDNA. Expression was normalized to L. erythrorhizon glyceraldehyde 3-phosphate dehydrogenase (LeGAPDH) using primers from Zhao et al.89.
Shikonin extraction and quantification
The extraction of total shikonins was modified from Boehm et al.90. Briefly, a 4 mL sample of growth media from each hairy root line was sampled at day 3 after transfer to M9 and darkness and extracted with 4 mL of chloroform. The chloroform layer was separated and dried under a gentle stream of N2 at 40 °C. Base hydrolysis was performed on the remaining residue in the tube by adding 2 mL of 1 M NaOH and shaking for 1 h at room temperature. The solution was neutralized by adding 1 mL of 6 M HCl and vortexed. Shikonin was extracted by adding 3 mL of ethyl acetate by liquid–liquid extraction. The ethyl acetate layer was separated and dried under N2, dissolved in 250 µL methanol, and 20 µL was used for detection by high performance liquid chromatography with diode array detection (HPLC-DAD). The extraction procedure was performed in reduced light to minimize the photo-degradation of shikonin.
HPLC-DAD analyses were performed with an Agilent 1260 Infinity HPLC system (Agilent Technologies). Chromatographic separation of shikonin was achieved using a Zorbax SB-C18 column (4.6 × 250 mm, Agilent) kept at 25 °C. The mobile phase gradient started at 60% A (30:70 acetonitrile and water with 0.1% formic acid) and 40% B (30:70 isopropanol and acetonitrile with 0.1% formic acid) with 1 min hold and then linearly increased to 99% B over 15 min with a hold of 4 min, and then returned to 40% B from 19 to 20 min with a hold of 1 min. Shikonin eluted at 8.4 min and was detected at 520 nm by DAD. Instrument operation and data analysis steps were performed through the Agilent ChemStation software. Shikonin quantitation by DAD was done by running a linear range of 1.25, 2.5, 5, 10, and 20 nmol calibration standards, followed by a linear regression formula calculation. Differences between shikonin content in each line (n = 3–4 biological replicates) was analyzed using one-way ANOVA and the means were compared with Tukey’s HSD post hoc test at 95% significant level.
For analysis of shikonins from L. erythrorhizon root periderm and vascular tissues for RNA-seq, HPLC coupled with fluorescence detection (HPLC-FLD; Fig. S5), chromatographic separation was conducted using a Zorbax Eclipse XDB-C18 column (4.6 × 150 mm, Agilent). The column was eluted at 25 °C using a linear gradient starting from 70% A (water with 0.1% formic acid) and 30% B (acetonitrile with 0.1% formic acid), to 1% A and 99% B over 40 min at a flow rate of 0.5 mL min−1, followed by a 10 min re-equilibration step. Shikonin eluted at 25.9 min and was detected by fluorescence using λex = 228 nm and λem = 390 nm after passing through an in-line post-column dry reactor packed with zinc dust, which was previously used for detection of the 1,4-naphthoquinone juglone51.
Data availability
Raw sequencing reads used for de novo whole-genome assembly and the final genome have been deposited in the Sequence Read Archive database under access number PRJNA596998.
References
Papageorgiou, V. P., Assimopoulou, A. N., Couladouros, E. A., Hepworth, D. & Nicolaou, K. C. The chemistry and biology of alkannin, shikonin, and related naphthazarin natural products. Angew. Chem. Int. Ed. 38, 270–300 (1999).
Skoneczny, D. et al. Metabolic profiling and identification of shikonins in root periderm of two invasive Echium spp. weeds in Australia. Molecules 22, 330 (2017).
Zhu, X. et al. Identification and localization of bioactive naphthoquinones in the roots and rhizosphere of Paterson’s curse (Echium plantagineum), a noxious invader. J. Exp. Bot. 67, 3777–3788 (2016).
Schmid, H. V. & Zenk, M. H. p-hydroxybenzoic acid and mevalonic acid as precursors of the plant naphthoquinone alkannin. Tetrahedron Lett. 12, 4151–4155 (1971).
Widhalm, J. R. & Rhodes, D. Biosynthesis and molecular actions of specialized 1,4-naphthoquinone natural products produced by horticultural plants. Hortic. Res 3, 16046 (2016).
Chen, X. et al. Shikonin, a component of Chinese herbal medicine, inhibits chemokine receptor function and suppresses human immunodeficiency virus type 1. Society 47, 2810–2816 (2003).
Duru, N., Gernapudi, R. & Zhou, Q. Chemopreventive activities of shikonin in breast cancer. Biochem Pharm. 3, e163 (2014).
Yazaki, K. Lithospermum erythrorhizon cell cultures: present and future aspects. Plant Biotechnol. 34, 131–142 (2017).
Gaisser, S. & Heide, L. Inhibition and regulation of shikonin biosynthesis in suspension cultures of Lithospermum. Phytochemistry 41, 1065–1072 (1996).
Boehm, R., Sommer, S., Li, S. M. & Heide, L. Genetic engineering on shikonin biosynthesis: expression of the bacterial ubiA gene in Lithospermum erythrorhizon. Plant Cell Physiol. 41, 911–919 (2000).
Köhle, A. et al. High level expression of chorismate pyruvate-lyase (UbiC) and HMG-CoA reductase in hairy root cultures of Lithospermum erythrorhizon. Plant Cell Physiol. 43, 894–902 (2002).
Wang, R. et al. An efficient multigram synthesis of alkannin and shikonin. Eur. J. Org. Chem. 2012, 1373–1379 (2012).
Wang, F., Yao, X., Zhang, Y. & Tang, J. Synthesis, biological function and evaluation of Shikonin in cancer therapy. Fitoterapia 134, 329–339 (2019).
Wang, S., Ping, L. & Teng, G. Different secondary metabolic responses to MeJA treatment in shikonin-proficient and shikonin-deficient cell lines from Arnebia euchroma (Royle) Johnst. Plant Cell Tiss. Org. 119, 587–598 (2014).
Zhu, Y. et al. Involvement of LeMDR, an ATP-binding cassette protein gene, in shikonin transport and biosynthesis in Lithospermum erythrorhizon. BMC Plant Biol. 17, 1–10. (2017).
Wu, F.-Y. et al. Transcriptome analysis explores genes related to shikonin biosynthesis in Lithospermeae plants and provides insights into Boraginales? evolutionary history. Sci. Rep. 7, 4477 (2017).
Takanashi, K. et al. Comparative proteomic analysis of Lithospermum erythrorhizon reveals regulation of a variety of metabolic enzymes leading to comprehensive understanding of the shikonin biosynthetic pathway. Plant Cell Physiol. 60, 19–28 (2019).
Wang, S. et al. CYP76B74 catalyzes the 3”-hydroxylation of geranylhydroquinone in shikonin biosynthesis. Plant Physiol. 179, 402–414 (2019).
Rai, A. et al. De novo transcriptome assembly and characterization of lithospermum officinale to discover putative genes involved in specialized metabolites biosynthesis. Planta Med. 84, 320–934. (2018).
Tang, C., Li, S., Wang, Y.-T. & Wang, X. Comparative genome/transcriptome analysis probes Boraginales’ phylogenetic position, WGDs in Boraginales, and key enzyme genes in the alkannin/shikonin core pathway. Mol. Ecol. Resour. 20, 228–241 (2020).
Ye, C., Hill, C. M., Wu, S., Ruan, J. & Ma, Z. DBG2OLC: Efficient assembly of large genomes using long erroneous reads of the third generation sequencing technologies. Sci. Rep. 6, 1–9 (2016).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
El-Gebali, S., et al. The Pfam protein families database in 2019. Nucleic Acids Res. https://doi.org/10.1093/nar/gky995 (2019).
Gene Ontology Consortium. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res 32, 258D–261D (2004).
Caspi, R., et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx935 (2018).
Waterhouse, R. M. et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548 (2018).
Wisecaver, J. H., Alexander, W. G., King, S. B., Todd Hittinger, C., Rokas, A. Dynamic evolution of nitric oxide detoxifying flavohemoglobins, a family of single-protein metabolic modules in bacteria and eukaryotes. Mol. Biol. Evol. https://doi.org/10.1093/molbev/msw073 (2016).
Emms, D. M. & Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 1–14. (2015).
Leebens-Mack, J. H. et al. One thousand plant transcriptomes and the phylogenomics of green plants. Nature https://doi.org/10.1038/s41586-019-1693-2 (2019).
Chase. M. W. et al. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. https://doi.org/10.1111/boj.12385 (2016).
Okada, K. et al. The AtPPT1 gene encoding 4-hydroxybenzoate polyprenyl diphosphate transferase in ubiquinone biosynthesis is required for embryo development in Arabidopsis thaliana. Plant Mol. Biol. 55, 567–577 (2004).
Yazaki, K., Kunihisa, M., Fujisaki, T. & Sato, F. Geranyl diphosphate:4-hydroxybenzoate geranyltransferase from Lithospermum erythrorhizon: cloning and characterization of a key enzyme in shikonin biosynthesis. J. Biol. Chem. 277, 6240–6246 (2002).
Yazaki, K., Fukui, H. & Tabata, M. Isolation of the intermediates and related metabolites of shikonin biosynthesis from Lithospermum erythrorhizon cell cultures. Chem. Pharm. Bull. (Tokyo) 34, 2290–2293 (1986).
Heide, L. & Tabata, M. Geranylpyrophosphate: p-hydroxybenzoate geranyltransferase activity in extracts of Lithospermum erythrorhizon cell cultures. Phytochemistry 26, 1651–1655 (1987).
Singh, R. S. et al. Expression of 3-hydroxy-3-methylglutaryl-CoA reductase, p-hydroxybenzoate-m-geranyltransferase and genes of phenylpropanoid pathway exhibits positive correlation with shikonins content in arnebia [Arnebia euchroma (Royle) Johnston]. BMC Mol. Biol. 11, 88 (2010).
Wu, S. J. S.-J. et al. Nitric oxide regulates shikonin formation in suspension-cultured onosma paniculatum cells. Plant Cell Physiol. 50, 118–128 (2009).
Wu, F. Y. et al. Transcriptome analysis explores genes related to shikonin biosynthesis in Lithospermeae plants and provides insights into Boraginales’ evolutionary history. Sci Rep. https://doi.org/10.1038/s41598-017-04750-1 (2017).
Kusano H., et al. Evolutionary developments in plant specialized metabolism, exemplified by two transferase families. Front Plant Sci. https://doi.org/10.3389/fpls.2019.00794 (2019).
Lyons E., Freeling M. How to usefully compare homologous plant genes and chromosomes as DNA sequences. Plant J. https://doi.org/10.1111/j.1365-313X.2007.03326.x (2008).
Zhang, W. J. et al. Expression analysis of shikonin-biosynthetic genes in response to M9 medium and light in Lithospermum erythrorhizon cell cultures. Plant Cell Tissue Organ Cult. 101, 135–142 (2010).
Ohara, K., Muroya, A., Fukushima, N. & Yazaki, K. Functional characterization of LePGT1, a membrane-bound prenyltransferase involved in the geranylation of p-hydroxybenzoic acid. Biochem J. 421, 231–241 (2009).
Ohara, K., Mito, K. & Yazaki, K. Homogeneous purification and characterization of LePGT1—a membrane-bound aromatic substrate prenyltransferase involved in secondary metabolism of Lithospermum erythrorhizon. FEBS J. 280, 2572–2580 (2013).
Lopez-Nieves S., et al. Relaxation of tyrosine pathway regulation underlies the evolution of betalain pigmentation in Caryophyllales. New Phytol. https://doi.org/10.1111/nph.14822 (2018).
Edger, P. P. et al. The butterfly plant arms-race escalated by gene and genome duplications. Proc. Natl Acad. Sci. USA 112, 8362–8366 (2015).
Moghe, G. D., Leong, B. J., Hurney, S. M., Jones, A. D. & Last, R. L. Evolutionary routes to biochemical innovation revealed by integrative analysis of a plant-defense related specialized metabolic pathway. Elife 6, e28468 (2017).
Loscher, R. & Heide, L. Biosynthesis of p-hydroxybenzoate from p-coumarate and p-coumaroyl-coenzyme A in cell-free extracts of lithospermum erythrorhizon cell cultures. Plant Physiol. 106, 271–279 (1994).
Block, A. et al. The origin and biosynthesis of the benzenoid moiety of ubiquinone (coenzyme Q) in Arabidopsis. Plant Cell 26, 1938–1948 (2014).
Soubeyrand, E. et al. The peroxidative cleavage of kaempferol contributes to the biosynthesis of the benzenoid moiety of ubiquinone in plants. Plant Cell 30, 2910–2921 (2018).
White, M. D. et al. UbiX is a flavin prenyltransferase required for bacterial ubiquinone biosynthesis. Nature https://doi.org/10.1038/nature14559 (2015).
Zhu, Y. et al. Involvement of LeMRP, an ATP-binding cassette transporter, in shikonin transport and biosynthesis in Lithospermum erythrorhizon. Plant Biol. 17, 1–9 (2017).
McCoy, R. M., Utturkar, S. M., Crook, J. W., Thimmapuram, J. & Widhalm, J. R. The origin and biosynthesis of the naphthalenoid moiety of juglone in black walnut. Hortic. Res. 5, 67 (2018).
Lindsey, I., Rivero, B. E., Calhoun, L. S., Grotewold, C. S. & Brkljacic, E. Standardized method for high-throughput sterilization of Arabidopsis seeds. J. Vis. Exp. 128, 56587 (2017).
Fujita, Y., Hara, Y., Ogino, T. & Suga, C. Production of shikonin derivatives by cell suspension cultures of Lithospermum erythrorhizon. Plant Cell Rep. 1, 59–60 (1981).
Wick, R. R., Judd, L. M. & Holt, K. E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 20, 1–10 (2019).
Simpson, J. T. et al. ABySS: A parallel assembler for short read sequence data. Genome Res. 19, 1117–1123 (2009).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE https://doi.org/10.1371/journal.pone. 0112963 (2014).
Vurture, G. W. et al. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997.
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, H. Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110 (2016).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: Allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinforma. 19, 1–10. (2018).
Holt C., Yandell M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinforma. https://doi.org/10.1186/1471-2105-12-491 (2011).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Korf, I. Gene finding in novel genomes. BMC Bioinforma. 5, 1–9 (2004).
Hoff, K. J., Lomsadze, A., Borodovsky, M., & Stanke M. in Gene Prediction: Methods and Protocols (ed Kollmar, M.). 65–95 (Springer New York: New York, NY, 2019).
Armenteros, J. J. A. et al. Detecting sequence signals in targeting peptides using deep learning. Life Sci. Alliance https://doi.org/10.26508/lsa.201900429 (2019).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2009).
Robinson, M. D., Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. https://doi.org/10.1186/gb-2010-11-3-r25 (2010).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995).
Bushnel, l B. BBTools Software Package (2017).
Haas, B. TransDecoder (Find Coding Regions within Transcripts) http://transdecoder.sourceforge.net (2012).
Stöver, B. C. & Müller K. F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinformatics https://doi.org/10.1186/1471-2105-11-7 (2010).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Kalyaanamoorthy, S., Minh,B. Q., Wong, T. K. F., Von Haeseler, A. & Jermiin L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. https://doi.org/10.1038/nmeth.4285 (2017).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. https://doi.org/10.1093/nar/gkz239 (2019).
Marçais, G. et al. MUMmer4: A fast and versatile genome alignment system. PLoS Comput Biol. https://doi.org/10.1371/journal.pcbi.1005944 (2018).
Karimi, M., Inzé, D. & Depicker, A. GATEWAY vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci. 7, 193–195 (2002).
Cui, W., Liu, W. & Wu, G. A simple method for the transformation of Agrobacterium tumefaciens by foreign DNA. Chin. J. Biotechnol. 11, 267–274 (1995).
Fang, R. et al. Transgenic analysis reveals LeACS-1 as a positive regulator of ethylene-induced shikonin biosynthesis in Lithospermum erythrorhizon hairy roots. Plant Mol. Biol. https://doi.org/10.1007/s11103-015-0421-z (2016).
Ghawana, S. et al. An RNA isolation system for plant tissues rich in secondary metabolites. BMC Res. Notes 4, 85 (2011).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25, 402–408 (2001).
Ye, J. et al. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinforma. 13, 134 (2012).
Lefever, S., Pattyn, F., Hellemans, J. & Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin. Chem. 59, 1470–1480 (2013).
Zhao, H. et al. Overexpression of LeMYB1 enhances shikonin formation by up-regulating key shikonin biosynthesis-related genes in Lithospermum erythrorhizon. Biol. Plant 59, 429–435 (2015).
Boehm, R., Sommer, S., Li, S.-M. & Heide, L. Genetic engineering on shikonin biosynthesis: expression of the bacterial ubiA gene in Lithospermum erythrorhizon. Plant Cell Physiol. 41, 911–919 (2000).
Acknowledgements
We thank Elena Yakubova and undergraduate students Haley Staniszewski and Michael McGill for their assistance in maintaining hairy root culture lines. This work was supported by start-up funds from Purdue University to J.H.W. and J.R.W., by a fellowship from the Anandamahidol Foundation (Thailand) to T.S., by the USDA National Institute of Food and Agriculture Predoctoral Grant 2018-67011-28032 to R.M.M., by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-1333468 to J.W.C., and by the NSF Dimensions of Biodiversity Program under Grant No. DEB-1831493 to J.H.W. This work was also supported by the USDA National Institute of Food and Agriculture Hatch Project numbers 177845 to J.R.W. and 1016057 to J.H.W.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Auber, R.P., Suttiyut, T., McCoy, R.M. et al. Hybrid de novo genome assembly of red gromwell (Lithospermum erythrorhizon) reveals evolutionary insight into shikonin biosynthesis. Hortic Res 7, 82 (2020). https://doi.org/10.1038/s41438-020-0301-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-020-0301-9
