
Abstract
Tea is one of the most popular nonalcoholic beverages due to its characteristic secondary metabolites with numerous health benefits. Although two draft genomes of tea plant (Camellia sinensis) have been published recently, the lack of chromosome-scale assembly hampers the understanding of the fundamental genomic architecture of tea plant and potential improvement. Here, we performed a genome-wide chromosome conformation capture technique (Hi-C) to obtain a chromosome-scale assembly based on the draft genome of C. sinensis var. sinensis and successfully ordered 2984.7 Mb (94.7%) scaffolds into 15 chromosomes. The scaffold N50 of the improved genome was 218.1 Mb, ~157-fold higher than that of the draft genome. Collinearity comparison of genome sequences and two genetic maps validated the high contiguity and accuracy of the chromosome-scale assembly. We clarified that only one Camellia recent tetraploidization event (CRT, 58.9–61.7 million years ago (Mya)) occurred after the core-eudicot common hexaploidization event (146.6–152.7 Mya). Meanwhile, 9243 genes (28.6%) occurred in tandem duplication, and most of these expanded after the CRT event. These gene duplicates increased functionally divergent genes that play important roles in tea-specific biosynthesis or stress response. Sixty-four catechin- and caffeine-related quantitative trait loci (QTLs) were anchored to chromosome assembly. Of these, two catechin-related QTL hotspots were derived from the CRT event, which illustrated that polyploidy has played a dramatic role in the diversification of tea germplasms. The availability of a chromosome-scale genome of tea plant holds great promise for the understanding of genome evolution and the discovery of novel genes contributing to agronomically beneficial traits in future breeding programs.
Similar content being viewed by others


Introduction
Tea, one of the most popular nonalcoholic beverages in the world, provides characteristic secondary metabolites, such as catechins, theanine, and caffeine that have numerous health benefits for humans1,2,3. The tea plant (Camellia sinensis (L.) O. Kuntze) originated in southwest China and has expanded worldwide to >50 countries4,5,6. By 2018, the worldwide cultivated area and production of tea have increased to 4.9 million hectares and 5.9 million tons, respectively7. Tea has created economic benefits and vast employment opportunities, especially in some Asian and African countries, such as China, India, Sri Lanka, and Kenya. In addition, due to distinctive sets of secondary metabolites (such as various catechins, caffeine, and theanine), tea has been widely applied in expounding the molecular mechanisms regulating catechins and theanine biosynthesis8,9,10,11,12. However, the lack of a high-quality genome sequence has become the main hindrance to gaining insights into secondary metabolite biosynthesis and fully understanding the evolution of tea plant. High levels of heterozygosity and repetitiveness pose a challenge to genome assembly in tea plant. Recently, two draft genomes of tea plant have been published by Illumina next-generation sequencing technology, but these genomes remain highly fragmented (scaffold N50: 449.5 kb in C. sinensis var. assamica genome and 1.4 Mb in C. sinensis var. sinensis (CSS) genome) and need improved completion13,14,15.
Chromosome-scale genome assembly is essential for genome-wide association study (GWAS) and the identification of quantitative trait loci (QTLs) governing important agronomic traits to facilitate gene cloning16. A high-quality reference genome also accelerates genome evolution involving ancient whole-genome duplications (WGDs), segmental duplications, tandem duplications, structural evolution, etc. The traditional BAC-by-BAC approach, integrating genetic and physical maps, has been used to produce high-quality assemblies17,18. However, this approach remains prohibitively expensive and laborious, which are bottlenecks for its widespread application to genome assembly. A genetic linkage map is another approach that assigns contigs or scaffolds to chromosomes19,20,21. A high-density SLAF-seq (SNP and SSR) genetic map of tea plant has been used to anchor scaffolds to chromosomes in the draft genome of CSS13,22. However, it is often difficult to generate a high-quality genome sequence, especially in centromeric regions, depending on the density of molecular markers, mapping population, and recombination events. Recently, Hi-C (high-throughput/resolution chromosome conformation capture), an effective and efficient approach, has been developed to guide genome assembly23,24,25. Due to the highly folded structures of chromosomes in a cell, sequences that are spatially proximal in three-dimensional (3D) space can be far apart along the linear chromosome. Hi-C detects spatially proximal DNA interactions by a proximity ligation method combined with Illumina sequencing. The frequency of contact between paired-end reads of loci can infer the distance of two DNA fragments, and further anchor and orient scaffolds. Hi-C has become a powerful approach for generating high-quality genome assemblies and has been widely performed in the genome assembly of animals and plants, such as humans24, Arabidopsis26, and black raspberry27.
WGD, also known as polyploidy, has been widely detected in plants and is considered an important evolutionary force in plants28,29. The WGD events are followed by chromosomal rearrangement, gene loss of most duplicates, and duplicate gene expression bias, causing a dramatic increase in species richness in angiosperms30,31. After the divergence of eudicots and monocots, a hexaploidization event (γ event) shared by all core eudicots occurred 140 million years ago (Mya)32. Because the Vitis vinifera genome had no additional WGD event except the hexaploidization event and preserved the ancestral eudicot chromosome structure, it has been widely used as a reference genome for the study of evolution33,34,35. Actinidia chinensis, the closest genus to Camellia in phylogenetic trees, showed two additional tetraploidization events after the hexaploidization event, namely, the Actinidia recent tetraploidization at ~18–20 Mya and the Actinidia ancient tetraploidization at ~50–57 Mya35,36. With the completion of the draft genome sequences of C. sinensis, two opinions regarding WGDs in the C. sinensis genome appeared: that two additional rounds of WGD events occurred in C. sinensis after the hexaploidization event13 and that only one additional WGD event occurred in C. sinensis independent with tetraploidization in A. chinensis14,36. Tandem duplicates that arise by unequal crossing over are closely adjacent identical genes in the same chromosome and are prevalent in eukaryotes37. Tandem duplicates usually tend to share common functions owing to co-regulated elements38. However, it has been revealed that some tandem duplicates have generated novel functions by acquiring novel transcription patterns39. Moreover, tandem duplicates play an important role in adaptive evolution to rapidly changing environments40.
In this study, Hi-C analysis of CSS ‘Shuchazao’ was performed to improve the draft genome sequence of CSS, generating a chromosome-scale assembly for tea plant. Based on the chromosome-scale genome, WGD and tandem duplication were identified to investigate recursive polyploidizations and diversification of duplicate genes. Furthermore, the published QTLs related to the catechins and caffeine content in tea were integrated into the chromosome-scale genome to facilitate the identification of the effective genes. These results improved our understanding of the evolution and diversification of duplicated genes in tea plant, laying a substantial foundation for the discovery of novel genes contributing to agronomically beneficial traits in future breeding programs.
Results
Chromosome-scale assembly of CSS
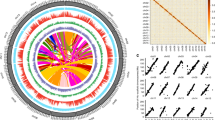
To obtain a chromosome-scale assembly of tea plant, a total of 1,126,108,661 Hi-C read pairs of CSS ‘Shuchazao’ (337.8 Gb, ~113-fold genome coverage) were generated and mapped to the draft genome of CSS13. After the removal of erroneous mappings and PCR duplicates, the remaining 507,043,204 read pairs (45.0% of total reads) were used to construct Hi-C linking information and a Hi-C scaffolding pipeline (misjoin correction, ordering, and orientation). Finally, the chromosome-scale assembly of the CSS V1.2 genome spanning 3.2 Gb of genome sequence was generated. In this study, the 2984.7 Mb genome sequence was clustered into 15 superscaffolds, accounting for 94.7% of the total genome size. The scaffold N50 of the CSS V1.2 genome was 218.1 Mb, ~157-fold higher than that of the draft genome (1.4 Mb, Supplementary Table 1). The Hi-C heatmap showed that 15 superscaffolds in the CSS V1.2 genome could be distinguished and perfectly represented 15 chromosomes (Fig. 1a). The chromosome names from chr1 to chr15 were defined by the previous genetic map from LG01 to LG15, respectively22. Of the 15 chromosomes, the longest and shortest assembled pseudomolecules were chr3 and chr15 at 264,061,170 and 139,796,614 bp, respectively, and the average length was 198,979,785 bp (Fig. 1b). To facilitate identification of functional genes, 32,311 protein-coding genes (95.2% of annotated genes in the draft genome) were anchored to the chromosomes of the CSS V1.2 genome. The number of genes on each chromosome varied significantly from 1467 to 3013 (mean, 2154) and showed that the gene number was correlated to chromosome size (r2 = 0.91, Pearson correlation coefficient). The size of transposable elements (TEs) had a higher coefficient of association (r2 = 0.99) than the number of genes. However, the proportion of TEs in each chromosome remained relatively constant, from 55.5% to 59.5% (mean, 57.4%, Supplementary Table 2).

a The heatmap represents the contact matrices generated by aligning the Hi-C data to the chromosome-scale assembly of the CSS V1.2 genome. b The length statistics of each chromosome of the CSS V1.2 genome resulting from the Juicer and 3D-DNA pipelines
To assess the improved genome, 6042 and 2380 available markers from the SLAF-seq22 and 2b-RAD41 genetic maps were assigned to the CSS V1.2 genome. After filtering multimapped sequences, 2035 and 1828 unambiguous alignments in the SLAF-seq and 2b-RAD genetic maps were retained, and 96.9% and 83.1% of these were correctly identified in the CSS V1.2 genome, respectively, in the sense that these markers appeared on the same chromosome in the genetic map and the CSS V1.2 genome (Supplementary Table 3). Collinearity comparison between the physical map and the genetic map validated the high contiguity and accuracy of the CSS V1.2 genome (Fig. 2).

a The y-axis represents the genetic positions of the SLAF-seq genetic map from the F1 population of CSS ‘Yingshuang’ and C. sinensis var. pubilimba ‘Beiyue Danzhu’. b The y-axis represents the genetic positions of the 2b-RAD genetic map from the F1 population of CSS ‘Longjing 43’ and CSS ‘Baihaozao’. The x-axis represents the physical positions of the chromosome-scale assembly of the CSS V1.2 genome
Whole-genome duplication
Two opinions, namely, that one or two additional WGD events in C. sinensis occurred after the hexaploidization event (γ event) shared by the eudicot genome, have been reported13,14. To clarify the question of how many rounds of WGD events occurred in C. sinensis, the sequence divergence of homologous genes and gene dotplots were used to infer ancient polyploidization events. First, self-comparison of the C. sinensis genome was carried out, and 254 homologous blocks containing 4748 gene pairs were identified in the intragenomic gene collinearity of C. sinensis. The synonymous nucleotide substitutions (Ks) of these homologous gene pairs were calculated. These homologous blocks resulted from multiple polyploidization events, so we classified them by the median Ks of each homologous block based on the phenomenon that Ks of homologous genes produced by one polyploidization event were usually similar. The histogram plot with the median Ks of homologous blocks showed two clearly distinguishable peaks (Supplementary Fig. 1), suggesting that two ancient polyploidization events exist in C. sinensis. The homologous blocks with relatively high Ks values might have resulted from the core-eudicot common hexaploidization event (ECH), while other blocks (Ks ≤ 0.7) might have been generated by the Camellia recent WGD event. The homologous gene dotplot within the C. sinensis genome integrated with the Ks information further clarified that one Camellia recent tetraploidization event (CRT) occurred after the ECH event (Supplementary Fig. 2). These blocks were classified into 177 ECH-related and 77 CRT-related homologous blocks, containing 2152 (45.3%, 12.2 gene pairs per block) and 2596 (54.7%, 33.7) gene pairs, respectively, revealing that CRT has a more highly conserved synteny than ECH in C. sinensis (Supplementary Table 4). To estimate the occurrence times of two polyploidization events, curve fitting of the Ks distribution of homologous genes resulting from the ECH and CRT events was performed. The peaks of the two polyploidization events were at 0.4 and 1.0, suggesting that the CRT and ECH events occurred at 58.9–61.7 and 146.6–152.7 Mya based on a neutral substitution rate of 3.39 × 10−936, respectively (Fig. 3a). Although the CRT event occurred near the divergence time of C. sinensis and A. chinensis from their common ancestor (61.2–65.3 Mya), a previous report indicates that the tetraploidization event was a lineage-specific WGD14,36 (Fig. 3b).

a Ks distribution between collinear genes within the C. sinensis genome or among genomes. CRT Camellia recent tetraploidization, ECH core-eudicot common hexaploidization, ART Actinidia recent tetraploidization, AAT Actinidia ancient tetraploidization. b Phylogenetic tree of C. sinensis, V. vinifera, and A. chinensis
To further verify two polyploidization events (ECH and CRT) in C. sinensis, the CSS V1.2 genome was compared with V. vinifera, known to be the closest relative to the eudicot ancestor structured. Based on the all-vs-all blastp analysis, 714 homologous blocks containing 14,831 gene pairs between C. sinensis and V. vinifera were identified (Supplementary Table 5). Using V. vinifera as the reference genome, for almost every V. vinifera chromosome, six collinear regions can be identified in the C. sinensis genome (Fig. 4). Among these, the best and second best-matched V. vinifera regions resulted from the CRT event and ECH event, respectively. Thus, this 1-to-2 relationship between V. vinifera and C. sinensis genomic regions inferred from the best-matching V. vinifera regions was a clear indication of a tetraploidization event in the C. sinensis genome after the split with V. vinifera. In addition, the following C. sinensis chromosome to V. vinifera chromosome correspondences were established (c for C. sinensis and v for V. vinifera as chromosome nomenclature): c1/v18-v8-v3-v6, c2/v1-v11-v15-v14-v3, c3/v13-v16-v14-v4-v7, c4/v5-v19-v12, c5/v10, c6/v3-v15-v14-v13-v12, c7/v8-v6-v7-v3-v12, c8/v11-v9-v17-v14-v7-v4, c9/v12-v19-v13-v16, c10/v5-v18, c11/v6-v7, c12/v8-v4-v1, c13/v2-v4-v11, c14/v1-v2-v4, c15/v9-v17. Among these, c5 was fused with its chromosomal segment duplications corresponding to v10. However, the fission and fusion of at least five chromosomes resulted in the longest chromosome c3.

The red, blue, and gray dots represent the best, secondary, and other matched homologous gene pairs resulting from the output of Blast software. The 19 chromosomes of the V. vinifera genome are colored by the seven eudicot ancestral chromosomes. The orthologous regions were identified and marked by squares
Polyploidization events increased the complexity of the C. sinensis genome, including genome size, gene content, and gene expression. For example, C. sinensis chr5 mainly contained two intrachromosomal segmental duplications, suggesting that it fused with its homolog after the recent tetraploidization event, corresponding to chr10 in V. vinifera. Although 250 paralogous gene pairs exhibited substantially conserved synteny between chr5a (chr5:21.3–86.1 Mb) and chr5b (chr5:89.9–218.1 Mb), these genes accounted for only 25.4% and 35.6% of all genes in chr5a and chr5b, respectively, and most genes exhibited divergence between the two duplicates. Moreover, the segmental length of chr5b was twice that of chr5a, resulting from variation in the number of TEs during genome evolution and diversification (78.3 Mb TEs in chr5b and 36.4 Mb in chr5a, Supplementary Fig. 3a). Among these retained genes both in chr5a and chr5b, expression divergence between duplicate genes was widespread during evolution, and 50.5–57.8% of duplicate genes had differential expression in one of the eight tissues (Supplementary Fig. 3b). The duplicate genes of asymmetrical evolution in sequence, structure, and expression may be important for the evolution of the C. sinensis genome.
Tandem gene duplication
Tandem duplication was also detected by BLASTP (e-value < 1e−20), with a maximum of five intervening genes. A total of 3262 tandem expanded regions were detected in the C. sinensis genome and were distributed unevenly on 15 chromosomes (Fig. 5a). The tandem expanded regions contained 9243 genes (28.6% of all genes on the chromosomes), which was higher than the numbers in A. chinensis (3111, 10.1%) and V. vinifera (5088, 21.5%, Supplementary Table 6). The most tandem duplicates in the C. sinensis genome were the result of a steady and unusually high rate of tandem duplicate gain after the recent tetraploidization event (Supplementary Fig. 4). The tandem genes had significantly lower expression levels than the nontandem genes in each of eight tissues (apical bud, young leaf, mature leaf, old leaf, young stem, root, flower, and fruit), and 26.5% of tandem genes were not expressed (FPKM < 1) in all tissues (Fig. 5c). These results revealed that the recently expanded or lineage-specific genes had lower expression, consistent with a previous report that gene expression was positively correlated with the age of gene occurrence42.

a C. sinensis genome topography and synteny. I represents the duplicated gene blocks within C. sinensis that were produced by ECH (green) and CRT (orange) events. II represents the frequency distribution of tandem genes in 1-Mb intervals across the 15 chromosomes, MAX = 22. III represents gene density ranging from 0 to 37 genes per Mb. b Identification of tandemly duplicated NBS-encoding genes. c Comparison of gene expression in eight tissues between tandem and nontandem NBS-encoding genes. d Comparison of the highly expressed apical genes between tandem and nontandem genes among the enriched genes involved in phenylpropanoid biosynthesis and flavonoid biosynthesis. e Phylogenetic tree and gene expression in eight tissues of six F3ʹ5ʹH genes belonging to two clusters
Furthermore, KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichment analysis of tandem duplication indicated that most of these genes were related to plant–pathogen interaction (ko04626), MAPK signaling pathway (ko04016), phenylpropanoid biosynthesis (ko00940), flavonoid biosynthesis (ko00941), anthocyanin biosynthesis (ko00942), isoflavonoid biosynthesis (ko00943), etc. (Supplementary Table 7). The tandem duplication retained in a lineage-specific fashion participated in abiotic and biotic stress tolerance, suggesting that tandem duplication played an important role in adaptive evolution to rapidly changing environments and/or interaction with pathogens40. More than 72.5% of the nucleotide binding site (NBS) genes, an important resistance (R) gene family, occurred in tandem duplication. These NBS genes could be divided into six classes according to their conserved domains: N, NL, CNL, CN, TN, and RN. The majority were N type and contained only the NB-ARC domain (Fig. 5b). Expression analysis in eight tissues, a measurement of the activity and function of genes, showed that NBS genes were highly or preferentially expressed in the root (Fig. 5c), resulting from the greater pressure on the root than the other tissues43. Among these NBS genes, the tandem duplication did not show lower expression levels, which was inconsistent with the overall result. These highly expressed tandem duplicates of NBS genes might increase C. sinensis resistance to dynamic environments and pathogens. Catechin, a type of flavonoid (flavan-3-ol), is synthesized through phenylpropanoid and flavonoid biosynthesis44,45. KEGG analysis indicated that five categories of KEGG orthology (COMT, HCT, CCR, CAD, and E1.14.11.61) involved in phenylpropanoid biosynthesis and six (DFR, CHS, LAR, CYP75A, FLS, and C12RT1) involved in flavonoid biosynthesis were significantly enriched in tandem duplicates. Structural genes involved in the two pathways with high expression in leaves generally played important roles in governing catechin contents, such as CsF3H, CsANS, and CsF3ʹ5ʹH45,46. Most highly expressed genes in the apical buds of these orthologs occurred in tandem duplication except for CAD, CCR, and FLS (Fig. 5d). For example, six F3ʹ5ʹH genes belonging to CYP75A that could regulate catechin contents and the ratio of di/tri-hydroxylated catechins were identified in two clusters (chr11:134.5–135.1 Mb and chr7:43.7–43.8 Mb). Of these, the highly expressed TEA013315.1 and TEA026296.1 have been previously cloned as CsF3ʹ5ʹHa and CsF3ʹ5ʹHb, respectively47 (Fig. 5e). It has been demonstrated that TEA013315.1 plays a crucial role in the concentration of catechins and could explain the variation in catechin contents among tea germplasms46. Tandem duplication increased functionally divergent genes that play important roles in tea-specific biosynthesis or stress response through sub- or neofunctionalization of the retained tandem duplicates, especially through evolution of gene expression.
Anchored QTL map
Catechin and caffeine, major secondary metabolites in young leaves of tea, contribute to the tea flavor and nutrient content. Among the catechins in green tea, (−)-epigallocatechin-3-gallate (EGCG) is the most abundant, followed by (−)-epigallocatechin (EGC), (−)-epicatechin-3-gallate (ECG), and (−)-epicatechin (EC)48,49. Based on previously reported QTLs related to the catechins and caffeine content in tea, 64 catechin- and caffeine-related QTLs were anchored to the CSS V1.2 genome (Fig. 6, Supplementary Table 8). Among these, eight caffeine-related QTLs were distributed in chr1, chr2, chr3, chr10, and chr11. The poor reproducibility of the caffeine-related QTLs identified in different experiments might be caused by the different parents used for the mapping populations. The distribution of catechin-related QTLs indicated that the EC-, ECG-, EGC-, and EGCG-related QTLs were closely linked in some regions. Six catechin-related QTL hotspots were detected on chr3, chr6, chr7, chr10, chr11, and chr15. The QTL hotspots in chr7 and chr11 showed high phenotypic variance explained (PVE > 20%). However, two QTL hotspots were not stable between the two populations of CSS ‘Yingshuang’ × C. sinensis var. pubilimba ‘Beiyue Danzhu’ and ‘TRFK 303/577’ × ‘GW Ejulu’. The analysis of WGD events implied that the regions of two hotspots were generated by the CRT event (Fig. 3, Supplementary Fig. 2). These results revealed that the QTL hotspots in chr11 might play an essential role in catechin diversification of Chinese tea germplasms, but chr7 might be responsible for tea germplasms in South Africa. In the QTL hotspot of chr11, the F3ʹ5ʹH gene TEA013315.1 has been cloned and validated to govern catechin traits in tea plant and its relatives. The tea genome sequence will provide the basis for the polymorphic markers, genes, and repeats within these QTL regions and facilitate the identification of their effective genes.

CAF caffeine content, C catechin, EC epicatechin, ECG epicatechin gallate, EGC epigallocatechin, EGCG epigallocatechin gallate. The arrow represents QTLs with high phenotypic variance explained (PVE > 20%)
Discussion
The first chromosome-scale genome of a highly heterozygous tea plant was successfully accomplished by Hi-C technology. The scaffold N50 of our assembly has been increased to 218.1 Mb, which is significantly higher than that of the draft genome. Moreover, collinearity comparison with the genetic map revealed that the chromosome-scale genome presented high contiguity and accuracy of assembly. Our results demonstrated that Hi-C technology could provide a rapid, suitable, and inexpensive approach to generate a high-quality chromosome-scale assembly compared with traditional approaches, such as BAC-by-BAC and genetic maps. These traditional approaches are confronted with many challenges when used for assembly of rather complex genomes (large genome size, polyploid, and high heterozygosity) because they are laborious and expensive. In contrast, Hi-C technology has overcome the existence insufficiency and become broadly available for many complex species50,51. Although Hi-C technology has accelerated the completion of chromosome-scale assembly of tea plants, our assembly is not perfect due to some misjoins of small adjacent contigs and missing bases. In the future, additional high-coverage PacBio long reads will be applied to correct the misjoins and fill the gaps.
Through the analysis of WGD events in the C. sinensis genome, we clarified that only one recent tetraploidization event (CRT, 58.9–61.7 Mya) occurred after the ancient hexaploidization event (ECH, 146.6–152.7 Mya) shared by the eudicot genome. A phylogenetic tree of homologous genes was constructed, and it was inferred that the CRT event occurred after the divergence of C. sinensis and A. chinensis from their common ancestor (61.2–65.3 Mya)14,36. After polyploidization events, the genomic architecture was surprisingly varied, including massive genomic rearrangements, homologous exchange, gene loss of duplicates, proliferation of TEs, and gene expression bias of duplicates, which play an important role in the formation and evolution of species. Small RNAs, as mediators of interactions in duplicated genomes, have influences on genomic architecture that reflect their functional roles31. Understanding the fundamental genomic structure accelerates plant improvement and utilization. For example, in maize, ~50% phenotype- or function-associated variation is more likely to have paralogs derived from the polyploidization event than singleton genes, suggesting that gene duplication followed by neofunctionalization or subfunctionalization plays an essential role in phenotypic variation52. Our observation that extraordinary variation in genome size, gene loss, gene expression, and tandem duplicates burst after the CRT event. Two QTL hotspots in chr11 and chr7 derived from the CRT event might be responsible for catechin diversification of the tea germplasms.
Tea is one of the most popular nonalcoholic beverages in the world and has numerous health benefits for humans. The availability of a chromosome-scale genome holds great promise for the understanding of fundamental genomic architecture and evolution, and for the improvement of tea plant quality (such as catechin, theanine and caffeine) and yield. Integrated with GWAS, map-based clone, and bulked segregant analysis, the chromosome-scale genome will accelerate the identification of trait-related variations or genes that can be used for the rapid, effective, and inexpensive selection of available germplasms or cultivars.
Materials and methods
Hi-C sequencing and genome sequence
Fresh leaves of CSS ‘Shuchazao’ grown at the China National Germplasm Tea Repository (Hangzhou, Zhejiang, China) were selected for Hi-C sequencing. An in situ Hi-C library based on DpnII was constructed as described previously53 and the resulting library with an insert size of ~300 bp was sequenced on a HiSeq 4000 under 2 × 150 bp mode. The draft genome sequence of CSS ‘Shuchazao’ was downloaded from the Tea Plant Information Archive (TPIA, http://tpia.teaplant.org/)13. The gene annotation and TE annotation used in this study were also obtained from the draft genome sequence of CSS ‘Shuchazao’13.
Chromosome assembly with Hi-C
Raw Hi-C reads were mapped to the draft genome sequence using bwa (version 0.7.17-r1188)54, and then erroneous mappings (MAPQ = 0) and duplicates were filtered by the Juicer pipeline55. The output of the Juicer pipeline was used for 3D-DNA56 analysis with default parameters, including misjoin correction, ordering, and orientation. To ensure the accuracy of assembly, the scaffold misjoins, ordering, and orientation were further checked and corrected manually based on the interaction matrix of scaffolds from the Juicebox visualization system57.
Comparison of genetic map and physical map
To assess the CSS V1.2 genome, two public genetic maps22,41 were used for collinearity analysis compared with the chromosome-scale assembly. A total of 6042 valid SNP markers of a SLAF-seq genetic map (F1 population, CSS ‘Yingshuang’ × C. sinensis var. pubilimba ‘Beiyue Danzhu’)22 and 2380 available markers of a 2b-RAD genetic map (F1 population, CSS ‘Longjing 43’ × CSS ‘Baihaozao’)41 were aligned to the assembled genome by Bowtie (version 1.2.1.1)58 with ‘-a –v 1’ parameters. To obtain unambiguous alignments, reads that mapped to more than two places in the genome were removed. The dotplot of collinearity comparison was drawn by Perl script with the SVG module.
Analysis of WGD and tandem duplication
Genes anchored to chromosomes in the C. sinensis, V. vinifera, and A. chinensis genomes were used to analyze genome evolution. First, protein sequences of C. sinensis were searched against the three genomes to find potentially homologous genes using BLASTP59 (e-value threshold, 1e−5). Second, large gene families with >30 matches were filtered out, and the remaining homologous genes were used for inferring homologous blocks by MCScanX60 software with ‘-s 4 –m 50 –w 5’ parameters. The synonymous nucleotide substitution rates (Ks) were calculated by using add_ka_and_ks_to_collinearity.pl implemented in MCScanX.
To distinguish the event-related homologous blocks, homologous blocks between the C. sinensis and V. vinifera genomes attributable to WGD events occurring after the tea-grape split were identified by the criterion that the best-matched homologous gene pairs accounted for >50% in a homologous block. To identify the ECH and CRT events in the C. sinensis genome, any homologous block with median Ks > 0.7 was defined as an ECH event, and any other was defined as a CRT event. The smooth curve of Ks distribution was obtained by using the Gaussian kernel function.
To identify tandem duplications, all genes anchored to the chromosomes were compared by BLASTP to find homologous genes (e-value < 1e−20). The homologous genes with a maximum of five intervening genes were defined as tandem duplicates61. KEGG enrichment analysis of tandem duplicates was performed by clusterProfiler package62 with the cutoff set at adjusted P value < 0.05.
RNA-seq data analysis
The RNA-seq data of eight tissues (apical bud, young leaf, mature leaf, old leaf, young stem, root, flower, and fruit) of CSS ‘Shuchazao’ were obtained from the NCBI SRA database submitted by Wei et al.13 (Supplementary Table 9). The raw Illumina reads were trimmed by Trimmomatic (v 0.36)63 and then aligned to the CSS V1.2 reference genome using HISAT264 with default settings. Differential expression between duplicate genes was calculated by the edgeR package65 with an FDR < 0.05 and at least a two-fold difference in expression levels.
Identification of NBS genes
All genes from the C. sinensis genome were annotated using HMMER 3.1b2 (http://hmmer.org/) against the Pfam database with an e-value threshold of 0.001. Genes with NB-ARC domains (PF00931) were defined as NBS genes and used for further analysis. These NBS genes were classified by different conserved domains identified by Pfam and the NCBI Conserved Domains Tool66. The conserved domains were confirmed using the following accession numbers: TIR (PF13676, PF01582), LRR (PF00560, PF07723, PF07725, PF12799, PF13306, PF13516, PF13504, PF13855, cl34836), and RPW8 (PF05659). CC domains were detected and confirmed by Paircoil267 with a P-score cutoff of 0.025.
Data availability
The raw Hi-C data of CSS ‘Shuchazao’ have been deposited in the NCBI Sequence Read Archive (SRA) under BioProject accession no. PRJNA596054. The CSS V1.2 assembly data is available at https://github.com/JiedanChen/TeaGenomeData.
References
Kanwar, J. et al. Recent advances on tea polyphenols. Front. Biosci. 4, 111–131 (2012).
Pervin, M. et al. Beneficial effects of green tea catechins on neurodegenerative diseases. Molecules 23, 1297 (2018).
Mancini, E. et al. Green tea effects on cognition, mood and human brain function: a systematic review. Phytomedicine 34, 26–37 (2017).
Chen, L., Apostolides, Z. & Chen, Z. M. Global Tea Breeding: Achievements, Challenges and Perspectives (University Press-Springer, Hangzhou, Zhejiang; 2012).
Chen, L., Yao, M. Z., Wang, X. C. & Yang, Y. J. Tea genetic resources in China. Int. J. Tea Sci. 8(2), 55–64 (2012).
Yao, M. Z., Ma, C. L., Qiao, T. T., Jin, J. Q. & Chen, L. Diversity distribution and population structure of tea germplasms in China revealed by EST-SSR markers. Tree Genet. Genomes 8, 205–220 (2011).
International Tea Committee. Annual Bulletin of Statistics (London, 2019).
Rani, A., Singh, K., Sood, P., Kumar, S. & Ahuja, P. S. p-Coumarate:CoA ligase as a key gene in the yield of catechins in tea [Camellia sinensis (L.) O. Kuntze]. Funct. Integr. Genomics 9, 271–275 (2009).
Singh, K. et al. An early gene of the flavonoid pathway, flavanone 3-hydroxylase, exhibits a positive relationship with the concentration of catechins in tea (Camellia sinensis). Tree Physiol. 28, 1349–1356 (2008).
Rani, A., Singh, K., Ahuja, P. S. & Kumar, S. Molecular regulation of catechins biosynthesis in tea [Camellia sinensis (L.) O. Kuntze]. Gene 495, 205–210 (2012).
Pang, Y. et al. Functional characterization of proanthocyanidin pathway enzymes from tea and their application for metabolic engineering. Plant Physiol. 161, 1103–1116 (2013).
Deng, W. W., Ogita, S. & Ashihara, H. Ethylamine content and theanine biosynthesis in different organs of Camellia sinensis seedlings. Z. Naturforsch. C. J. Biosci. 64, 387–390 (2009).
Wei, C. et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl Acad. Sci. USA 115, E4151–E4158 (2018).
Xia, E. H. et al. The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis. Mol. Plant 10, 866–877 (2017).
Xia, E. et al. The tea plant reference genome and improved gene annotation using long-read and paired-end sequencing data. Sci. Data 6, 122 (2019).
Philippe, R. et al. A high density physical map of chromosome 1BL supports evolutionary studies, map-based cloning and sequencing in wheat. Genome Biol. 14, R64 (2013).
International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 436, 793–800 (2005).
Schnable, P. S. et al. The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115 (2009).
Prochnik, S. et al. The cassava genome: current progress, future directions. Tropical Plant Biol. 5, 88–94 (2012).
Jia, J. et al. Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496, 91–95 (2013).
Wang, S. et al. Sequence-based ultra-dense genetic and physical maps reveal structural variations of allopolyploid cotton genomes. Genome Biol. 16, 108 (2015).
Ma, J. Q. et al. Large-scale SNP discovery and genotyping for constructing a high-density genetic map of tea plant using Specific-Locus Amplified Fragment Sequencing (SLAF-seq). PLoS ONE 10, e0128798 (2015).
Kaplan, N. & Dekker, J. High-throughput genome scaffolding from in vivo DNA interaction frequency. Nat. Biotechnol. 31, 1143–1147 (2013).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125 (2013).
Dekker, J., Marti-Renom, M. A. & Mirny, L. A. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat. Rev. Genet. 14, 390–403 (2013).
Xie, T. et al. De novo plant genome assembly based on chromatin interactions: a case study of Arabidopsis thaliana. Mol. Plant 8, 489–492 (2015).
Jibran, R. et al. Chromosome-scale scaffolding of the black raspberry (Rubus occidentalis L.) genome based on chromatin interaction data. Hortic. Res. 5, 8 (2018).
Qiao, X. et al. Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 20, 38 (2019).
Jiao, Y. et al. Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100 (2011).
Soltis, D. E. et al. Polyploidy and angiosperm diversification. Am. J. Bot. 96, 336–348 (2009).
Wendel, J. F., Jackson, S. A., Meyers, B. C. & Wing, R. A. Evolution of plant genome architecture. Genome Biol. 17, 37 (2016).
Moore, M. J., Bell, C. D., Soltis, P. S. & Soltis, D. E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl Acad. Sci. USA 104, 19363–19368 (2007).
Paterson, A. H. et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492, 423–427 (2012).
Wang, J. et al. An overlooked paleotetraploidization in Cucurbitaceae. Mol. Biol. Evol. 35, 16–26 (2018).
Wang, J. P. et al. Two likely auto-tetrapl oidization events shaped kiwi fruit genome and contributed to establishment of the Actinidiaceae family. iScience 7, 230–240 (2018).
Wu, H. et al. A high-quality Actinidia chinensis (kiwifruit) genome. Horticulture Res. 6, 117 (2019).
Freeling, M. Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 60, 433–453 (2009).
Li, X. et al. Genome-wide analysis of basic/helix-loop-helix transcription factor family in rice and Arabidopsis. Plant Physiol. 141, 1167–1184 (2006).
Fan, C., Chen, Y. & Long, M. Recurrent tandem gene duplication gave rise to functionally divergent genes in Drosophila. Mol. Biol. Evol. 25, 1451–1458 (2008).
Hanada, K., Zou, C., Lehti-Shiu, M. D., Shinozaki, K. & Shiu, S. H. Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Plant Physiol. 148, 993–1003 (2008).
Xu, L. Y. et al. High-density SNP linkage map construction and QTL mapping for flavonoid-related traits in a tea plant (Camellia sinensis) using 2b-RAD sequencing. BMC Genomics 19, 955 (2018).
Stein, J. C. et al. Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nat. Genet. 50, 285–296 (2018).
Fu, Y. et al. NBS-encoding genes in Brassica napus evolved rapidly after allopolyploidization and co-localize with known disease resistance loci. Front. Plant Sci. 10, 26 (2019).
Winkel-Shirley, B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 126, 485–493 (2001).
Guo, F., Guo, Y., Wang, P., Wang, Y. & Ni, D. Transcriptional profiling of catechins biosynthesis genes during tea plant leaf development. Planta 246, 1139–1152 (2017).
Jin, J. Q., Ma, J. Q., Yao, M. Z., Ma, C. L. & Chen, L. Functional natural allelic variants of flavonoid 3’,5’-hydroxylase gene governing catechin traits in tea plant and its relatives. Planta 245, 523–538 (2017).
Guo, L. et al. Functional analysis of flavonoid 3’-hydroxylase and flavonoid 3’,5’-hydroxylases from tea plant (Camellia sinensis), involved in the B-ring hydroxylation of flavonoids. Gene 717, 144046 (2019).
Balentine, D. A., Wiseman, S. A. & Bouwens, L. C. The chemistry of tea flavonoids. Crit. Rev. Food Sci. Nutr. 37, 693–704 (1997).
Shahidi, F. Antioxidants in food and food antioxidants. Nahrung 44, 158–163 (2000).
Zhang, J. et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 50, 1565–1573 (2018).
Bickhart, D. M. et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 49, 643–650 (2017).
Wallace, J. G. et al. Association mapping across numerous traits reveals patterns of functional variation in maize. PLoS Genet. 10, e1004845 (2014).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Myburg, A. A. et al. The genome of Eucalyptus grandis. Nature 510, 356–362 (2014).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Marchler-Bauer, A. et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229 (2011).
McDonnell, A. V., Jiang, T., Keating, A. E. & Berger, B. Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics 22, 356–358 (2006).
Acknowledgements
This research was supported by the Ministry of Agriculture of China through the Earmarked Fund for China Agriculture Research System (CARS-019), the Chinese Academy of Agricultural Sciences through the Agricultural Science and Technology Innovation Program (CAAS-ASTIP-2017-TRICAAS) to L.C., and the Zhejiang Provincial Natural Science Foundation of China (Grant LQ20C160010) to J.-D.C.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, JD., Zheng, C., Ma, JQ. et al. The chromosome-scale genome reveals the evolution and diversification after the recent tetraploidization event in tea plant. Hortic Res 7, 63 (2020). https://doi.org/10.1038/s41438-020-0288-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-020-0288-2
This article is cited by
-
Circadian rhythm response and its effect on photosynthetic characteristics of the Lhcb family genes in tea plant
BMC Plant Biology (2024)
-
Decoding the complete organelle genomic architecture of Stewartia gemmata: an early-diverging species in Theaceae
BMC Genomics (2024)
-
Chromosome-scale genome assembly of oil-tea tree Camellia crapnelliana
Scientific Data (2024)
