
Abstract
The cultivated petunia (Petunia ×hybrida) is derived from the progenitor species P. axillaris and P. integrifolia. The hybridization dates back only to the 1830s, though intensive breeding efforts have yielded cultivars exhibiting incredible diversity for many traits, including growth habit, flower color, and flower size. Until now, little is known about the genetic diversity and genomic background of modern cultivars. Here we selected a panel of 13 cultivars with contrasting growth habits and three wild species (the progenitors and P. exserta) to estimate the genomic contribution from the ancestral species and to study whether the variation of the genetic origin could be associated with different breeding programs or morphological variability. Transcriptome sequencing identified 1,164,566 SNPs representing 98.4% (32,451) of the transcripts that cover 99.2% (of 52,697,361 bp) of the P. axillaris transcriptome. Cultivars with an upright growth habit had more homozygous alleles and more P. axillaris-derived alleles than trailing cultivars, while mounded cultivars had intermediate heterozygosity. Unlike previous studies, we found the proportions of alleles derived from each progenitor species varied across cultivars but overall were not biased toward one progenitor species, suggesting diverse selection during cultivar development. For trailing cultivars, alleles potentially introgressed from other wild species (“out” alleles) were enriched. The “out” alleles were clustered in particular regions of chromosomes, suggesting that these regions may be hotspots of introgression. Transcripts in these regions were enriched with gene ontology terms associated with growth habit. This study provides novel insight into the contributions of progenitor species to the genomic background of modern petunia cultivars and identifies genome regions that may harbor genes conferring the trailing growth habit for further exploration.
Similar content being viewed by others

Introduction
The garden petunia, Petunia ×hybrida, consistently ranks among the top selling annual bedding plants in the U.S. and Europe. In 2018, petunia ranked first among annual bedding plants in the U.S., with a wholesale value of $141.7 million1. The genus Petunia contains 14 species found in Argentina, Bolivia, Brazil, and Uruguay2. Petunia is derived from crosses between P. axillaris and one or more species within the P. integrifolia clade2,3,4.
Domestication of the cultivated petunia is quite recent, with first mention of the hybrid occurring in 18353. Since then, commercial breeders have released hundreds of cultivars exhibiting considerable diversity for traits like flower color, pattern and size, and growth habit5. The breeding history has sometimes involved hybridization with additional crop wild relative species. For example, petunia cultivars traditionally exhibited an upright growth habit3. However, a major new form of plant architecture for petunia, a strongly prostrate (trailing) and radial growth habit, was introduced with the release of the cultivar ‘Wave Purple’. This novel growth habit was likely the result of introgression of the species P. altiplana into commercial germplasm, as this is the only Petunia species exhibiting this growth habit2. Following the release of ‘Wave Purple’ and a second cultivar with a prostrate growth habit, petunia ‘Surfinia Red’, a large number of cultivars exhibiting growth habits intermediate to the upright and trailing types (mounded and mounding/trailing types) began to appear on the market.
In recent years, there has been a considerable increase in sequence availability for Petunia spp., including genome sequences for P. axillaris and P. inflata (a close relative of P. integrifolia)5, and transcriptome sequences for P. axillaris, P. exserta, P. ×hybrida, and P. integrifolia6,7,8. In addition, we have generated high-density single-nucleotide polymorphism (SNP)-based genetic maps for two interspecific Petunia recombinant inbred line (RIL) populations, including a P. integrifolia × P. axillaris population9,10,11,12. While these resources will greatly facilitate the elucidation of the genetic control of important traits, sequence resources representing the diversity of commercial petunia cultivars are currently lacking.
Bombarely et al.5 utilized the genome sequences of P. axillaris and the close P. integrifolia relative P. inflata and transcriptome data from three petunia laboratory strains, ‘Mitchell’, R27, and R143, to determine that the contribution of P. axillaris varied from 49.8 to 62.1%. However, these laboratory strains are not very representative of commercial cultivars, as R27 and R143 have been inbred for many generations, and ‘Mitchell’ is a doubled haploid derived from a cross between P. axillaris and petunia ‘Rose of Heaven’13. The contributions of P. axillaris and P. integrifolia to the genetic background of modern commercial cultivars are largely unknown.
The objectives of this study were to generate transcriptome-derived SNPs from a broad panel of petunia cultivars to evaluate the genetic diversity of petunia cultivars representing several breeding companies and a broad range of phenotypes, assess the relative contributions of P. axillaris and P. integrifolia to the genomic composition of these cultivars, identify genomic regions that may be derived from other (“outgroup”) crop wild relative species, and to further identify potential associations between prostrate growth habit and outgroup-derived genomic loci.
Results
SNP detection, annotation, and validation
Paired-end sequencing of three petunia cultivars (‘Wave Purple’, ‘Surfinia Red’, and ‘Madness Yellow’) contained 85–109 million high-quality reads. The average depth of coverage for uniquely mapped reads ranged from 37.1 to 53.3. The 10 other cultivars yielded 12.2 to 25.8 million high-quality single-end reads (Table 1). Transcriptome data for P. axillaris, P. integrifolia, and P. exserta were previously described6.
A total of 1,353,141 SNPs were detected among the 16 genotypes (3 wild species and 13 cultivars). The SNPs were further filtered to exclude those where no polymorphisms were detected between the cultivars and the reference genotype (P. axillaris), which left 1,164,566 SNPs (entire dataset). These SNPs were located on 32,451 transcripts with a total length of 52,697,361 bp, which was 98.4% of the total transcripts and covered 99.2% of the P. axillaris transcriptome assembly length6. A total of 74,218 SNPs with no missing data (no missing) were detected among the 16 genotypes. This set of no missing SNPs were located on 7815 transcripts with a total length of 16,790,590 bp. They comprised 23.7% of the P. axillaris transcripts and covered 31.6% of the reported P. axillaris transcriptome6.
Within the entire SNP dataset, the SNP numbers varied greatly between cultivars (Table 2). For example, 61,959 SNPs were detected between ‘Vista Silverberry’ (average dp = 4.3) and ‘Orchid Daddy’ (average dp = 3.1), almost 3 times the number of SNPs between ‘Vista Silverberry’ and ‘Vista Bubblegum’ (23,040; average dp = 5). For cultivars, the number of SNPs was generally higher with P. integrifolia than with P. axillaris (Table 2). The number of SNPs between ‘Madness Yellow’ or ‘Fantasy Rose’ and P. integrifolia was almost twice that with P. axillaris. While for ‘Tidal Wave Silver’, ‘Vista Silverberry’, and ‘Vista Bubblegum’, similar number of SNPs were detected for the cultivars between both progenitor species (Table 2).
A total of 22 predicted SNPs were selected for validation. Sanger sequencing of the targeted amplified region showed that all of these predicted SNPs were detected. For the cultivars, 91.2% of these predicted SNPs were validated. The <9% mis-genotyped samples were mainly from heterozygous SNPs validated as homozygous. This normally happened for genotypes with low minor allele frequency. Overall, the SNP validation results showed high accuracy of the SNP prediction pipeline.
Detecting regions under selection for petunia cultivars
The 74,218 SNP loci with no missing data were tested for signatures of selection based on their allelic differentiation from the expected neutrality14. The test was carried out by BayeScan, which incorporated the uncertainty on allele frequency and is thus suitable for datasets with small sample sizes15. When the cultivars were considered as one population and each wild species considered separately, BayeScan did not detect any outliers with default parameters at the default false discovery rate. When only cultivars were used, still no outliers were found, which indicated no significant signature of selection for petunia cultivars.
The proportion of ancestral genomic contribution varies within petunia cultivars
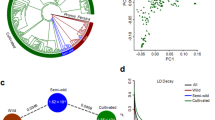
The transcriptome heterozygosity for each genotype was estimated using both the entire SNP dataset (1,164,566) and the no-missing SNP dataset (74,218) (Fig. 1). Both datasets showed that, among all 16 genotypes, P. integrifolia had the highest heterozygosity (59.3%) and P. axillaris and P. exserta had similar and the lowest heterozygosity. The cultivars had a range of heterozygosity rates sitting between the three wild species. In general, the trailing cultivars had higher transcriptome heterozygosity than the upright cultivars, and the mounded cultivars had heterozygosity rates in between. The low rates of transcriptome heterozygosity for the upright types (generally 10–15%) are particularly surprising, as most of these cultivars are F1 hybrids.

The heterozygosity rate is calculated as the number of heterozygous SNPs divided by the total number of SNPs. The cultivars and wild species were arranged according to the PCA similarity on PC1 (Supplementary Fig. 1).
The genetic background for each cultivar was further parsed (with 1,164,566 SNPs) according to similarity with the progenitor species P. axillaris and P. integrifolia following two methods: (1) the method of Bombarely et al.5 (approach 1); and (2) a two-model thresholds approach (approach 2) (Fig. 2). Following Bombarely et al.5, genetic dissection showed that the total percentage of homozygous SNPs ranged from 54.5% to 83.8% across the cultivars, whereas the total percentage of homozygous alleles derived from P. axillaris and P. integrifolia in these cultivars ranged from 34.0% to 53.3% (Fig. 2a).

The categories were derived from the genotypic similarity of the cultivars with P. axillaris, P. integrifolia, or other species represented by the percentage of transcriptome-derived SNPs in each genotype category. The estimation was made using the entire set of the SNPs (1,164,566 SNPs). a The estimated SNP proportion was calculated following Bombarely et al.5. b The estimated SNP proportion using the “exact parents” model (a) and the “pure parents” model” (b). Out: No polymorphisms detected between P. axillaris and P. integrifolia, whereas the cultivar genotypes had a different allele at the locus. Unknown: Allele origin could not be determined due to missing genotyping data in either P. axillaris or P. integrifolia. The cultivars were arranged according to the PCA similarity on PC1 (Supplementary Fig. 1).
Most of the alleles in the cultivars were derived from the progenitor species. The majority of heterozygous SNPs in the cultivars were heterozygous alleles of P. axillaris/P. integrifolia (7.4–27.7%), followed by heterozygous SNPs with one nucleotide from the common allele of P. axillaris and P. integrifolia and another nucleotide other than the current P. axillaris or P. integrifolia, which were named “out” genotypes. The “out” genotypes (3.3–14.3%) could come from another species or from other P. axillaris and P. integrifolia genotypes than those used in this study.
A higher percentage of P. axillaris alleles (34.7–45.2%) were detected in the cultivars than P. integrifolia alleles (15.5–26.9%) when the number of P. axillaris and P. integrifolia alleles in the heterozygous P. axillaris/P. integrifolia SNPs were set as 50% of those in homozygous SNPs (P. axillaris/P. axillaris or P. integrifolia/P. integrifolia), and the loci where P. axillaris and P. integrifolia showed no polymorphisms were excluded (25.1–31.1%). The percentage of P. axillaris alleles was higher in the upright cultivars compared to the trailing types, while the percentage of P. integrifolia alleles was higher in the trailing cultivars compared with the upright cultivars.
Interestingly, alleles not derived from either progenitor species (“out” alleles) were detected across all cultivars, which indicates that there could be allele introgression events from other species during cultivar development (Fig. 2 and Supplementary Table 2). Those alleles may comprise 4.5–9.9% of the SNPs in the transcriptome. This estimation was based on the assumption that the heterozygous “out” SNPs [including P. axillaris/out, P. integrifolia/out, and (P. axillaris = P. integrifolia)/out] had 50% fewer “out” alleles compared to homozygous “out” SNPs (out/out or out1/out2). ‘Wave Purple’ and ‘Surfinia Red’ had the largest percentage of “out” alleles among these cultivars (~9.9%).
For the P. axillaris genotype used in this study, approximately 59% of the SNPs showed “P. axillaris only” alleles, and for the P. integrifolia sample, about 35% SNPs showed “P. integrifolia only” alleles. In addition, P. axillaris and P. integrifolia shared approximately 35% common homozygous alleles.
It has been reported that the first petunia hybrids were produced by multiple crosses from different accessions of the two parent clades2,16. The exact genotypes of the wild species from which the current petunia cultivars were derived are unknown. In this study, both P. axillaris and P. integrifolia accessions had some heterozygosity (the heterozygosity of P. integrifolia was high). Bombarely et al.5 assumed that both parental accessions were 100% homozygous, which probably will not accurately represent the true genetic components of the cultivars. Therefore, a “two-threshold” method was implemented to estimate the range of the genetic proportion in the cultivars derived from the progenitor species. To further estimate the range of genomic components from the progenitor species, we developed a model assuming two “extreme statuses” of the P. axillaris and P. integrifolia genotypes: (1) the hypothetical progenitor species are 100% homozygous, with no common alleles (P. axillaris is 0/0 and P. integrifolia is 1/1, and no genomic similarity); and (2) the P. axillaris and P. integrifolia samples used in this study (with the current heterozygosity) are the exact progenitor genotypes from which these cultivars were developed (Fig. 2b). Under the first assumption, in the cultivars, the contribution of P. axillaris alleles ranged from 62.0% to 78.9%, and the contribution from P. integrifolia ranged from 20.8% to 37.6%. In addition, <1% SNPs had alleles that came from species other than P. axillaris or P. integrifolia (“out” alleles). Under the second assumption, 21.6–41.6% of the P. axillaris SNPs and 8.8–27.8% of P. integrifolia SNPs were detected in the cultivars. In addition, 16.8–28.9% of the SNPs were “out” alleles and 19.8–30.5% of the SNPs shared common alleles between P. axillaris and P. integrifolia. Under both assumptions, P. axillaris SNP alleles were estimated to comprise a higher percentage for the upright cultivars than the trailing cultivars while the P. integrifolia and “out” alleles were higher in the trailing than the upright cultivars. Also, under the second assumption, the estimated percentage of P. axillaris and P. integrifolia shared alleles was lower in the trailing cultivars than the upright types. The estimated ancestral alleles contribution from approach 1 (using the real dataset) is within the estimated progenitor proportion threshold using the hypothetical ancestor genotype model (approach 2).
The distribution of the ancestral alleles across the petunia genome
To dissect the genome-wide distribution of the ancestor origins for the cultivars, each locus was calculated for the percentage of similarity with the genotype of P. axillaris following Bombarely et al.5. The similarity ratio was then projected onto the high-density genetic linkage map according to the corresponding locations of the transcripts on the P. axillaris scaffolds and the genotyping-by-sequencing (GBS)-SNP locations on the AE (P. axillaris × P. exserta) genetic linkage map that we generated previously6,11. Overall, 1345 unique transcripts were placed on the AE map corresponding to 1978 GBS-SNP loci, which was 31.4% of the total SNPs on the AE map (Fig. 3). These 1345 transcripts had 65,170 SNP loci, and the percentage of similarity with P. axillaris was calculated for each transcript of each cultivar. A very small portion of transcripts (32 transcripts) were mapped to multiple locations on the AE map, yet most of these locations could be combined because of their short genetic distances (located within 1 cM). A few transcripts (<10) were mapped to locations >20 cM apart on the same linkage group. In this case, both locations were kept and the percentage of similarity with P. axillaris were compared with the adjacent mapped regions to gain a comprehensive understanding of the overall progenitor background distribution on the linkage map.

Ring 1 represents the genetic distance between markers on the AE map, while rings 2–17 represent the location and relative abundance of all P. axillaris alleles in: (2) P. axillaris, (3) P. integrifolia, (4) P. exserta, (5) Madness Yellow, (6) Storm Blue, (7) Super Cascade White, (8) Fantasy Rose, (9) Madness Red, (10) Orchid Daddy, (11) Supertunia Yellow, (12) Avalanche Salmon, (13) Tidal Wave Silver, (14) Wave Purple, (15) Surfinia Red, (16) Vista Bubblegum, and (17) Vista Silverberry. Dark red indicates highest similarity to P. axillaris while dark blue indicates lowest similarity to P. axillaris. The three “outer layers” are the three wild species. The cultivars were arranged, from outer to inner ring, according to their similarity with P. axillaris.
Some consistent patterns could be detected from the genome-wide distribution of genetic similarity with the hypothetical progenitor species P. axillaris (Fig. 3). P. axillaris had small regions of heterozygosity that tended to distribute toward the end of each linkage group. The P. integrifolia genotype had high heterozygosity and most regions on the linkage group were heterozygous. P. exserta had overall high similarity with P. axillaris. The regions with less identity were either heterozygous regions or regions with alleles introgressed from “out” (other) species or genotypes. We detected regions across the cultivar panel where both P. axillaris and P. integrifolia samples had high similarity with P. axillaris (red color), while all cultivars had low similarity (blue color). These could be the regions where the “out” alleles were introgressed.
No chromosome (linkage group) with exclusive genetic similarity with either of the progenitor species was detected for the cultivars. The percentage of similarity with P. axillaris was lower in the trailing cultivars compared with upright cultivars. Consistent with the principal component analysis (PCA) analysis (Supplementary Fig. 1), the cultivars ‘Vista Bubblegum’ and ‘Vista Silverberry’ showed almost identical ancestral genetic contribution pattern on the linkage maps, indicating a closely related breeding background.
The wild species introgression pattern in trailing and upright cultivars
SNPs between the representative trailing and upright cultivar groups were distributed on 95.8% of the P. axillaris transcriptome (839,794 SNPs located on 31,613 transcripts). For all the cultivars in the representative growth habit group, the proportion of each genotype category did not seem to change significantly in comparison with their proportions in the entire SNP set (Fig. 4a). Since neither P. axillaris nor P. integrifolia have a trailing growth habit, we assumed that the trailing growth habit was most likely contributed by the “out” alleles introgressed from another species. The “out” alleles are distributed in genotyping categories 6, 7, 8, 9, and 10. Of these, the vast majority (over 99.9%) were category 6 and 7 SNPs. When comparing the proportion of “enriched SNPs” (SNPs that are only polymorphic between the trailing and upright cultivar groups/total number of SNPs) in each genotyping category for each cultivar, the proportion of category 7 [heterozygous SNPs (P. axillaris = P. integrifolia)/out, Supplementary Table 1] was significantly increased only for the trailing cultivars, in comparison with the upright cultivars, and had an overall higher percentage than the expected average proportion (Fig. 4b). The enriched category 7 genotypes in the trailing cultivars could indicate that, in this category (genotyping pattern), alleles introgressed from other petunia species might be related to the horizontal growth habit of trailing petunia cultivars.

a Overview of the distribution of the percentage of SNPs between the trailing cultivars (‘Wave Purple’, ‘Surfinia Red’, and ‘Vista Bubblegum’) and the upright cultivars (‘Orchid Daddy’, ‘Strom Blue’, and ‘Madness Yellow’) dissected into each genotype category (Bombarely et al.5), compared with the percentage of SNPs in the entire dataset. b Comparison of the percentage in each genotype category using SNPs between the trailing cultivars (blue), the upright cultivars (orange), and the percentage of SNPs using the entire dataset (green). The genotyping categories were as in Fig. 2a. The cultivars on the x-axis were arranged according to their growth habit (trailing: ‘Wave Purple’, ‘Surfinia Red’, and ‘Vista Bubblegum’; upright: ‘Orchid Daddy’, ‘Strom Blue’, and ‘Madness Yellow’).
About 19.2% of the P. axillaris transcriptome (6323 transcripts) had SNPs that were polymorphic between the trailing and upright cultivars with alleles introgressed from a third species (“out” alleles from genotype category 6, 7, 8, 9, or 10). Of those, 2463 transcripts (7.5% of the P. axillaris transcriptome assembly) had category 7 SNPs in every representative trailing cultivar, while no category 7 SNPs were present in any upright cultivar. To characterize the genome-wide “third” species introgression patterns, which might elucidate regions correlated with the petunia trailing growth habit, we first placed transcripts that had any of the “out” genotype categories (genotype categories 6, 7, 8, 9, and 10) and were polymorphic between the trailing and upright cultivar groups on the AE linkage map (Fig. 5). Overall, 323 transcripts were placed on the AE linkage map. We then examined the regions where genotype 7 alleles were present only in the trailing cultivars, which could harbor genes related to the trailing growth habit. The Circos plot showed that the outgroup introgression regions were present on every chromosome. The location and abundance of introgressed “out” alleles on the AE map indicated that the introgressions tended to concentrate in high recombination frequency regions of the genome, and the abundance of overall “out” alleles and the 7 type-only alleles were consistent. The introgression frequency varied slightly among the three trailing type cultivars, with ‘Wave Purple’ and ‘Surfinia Red’ having overall both higher “out” and category 7 allele frequencies than ‘Vista Bubblegum’ (Fig. 5).

Ring 1 represents the genetic distance between markers on the AE map, while rings 2–10 represent the location and the frequency of introgressed alleles (SNPs) representing (2) all “out” alleles in the trailing group, (3) all category 7 [heterozygous (P. axillaris = P. integrifolia)/out] alleles in the trailing group, (4) all “out” alleles in Wave Purple, (5) all category 7 alleles in Wave Purple, (6) all “out” alleles in Surfinia Red, (7) all category 7 alleles in Surfinia Red, (8) all “out” alleles in Vista Bubblegum, (9) all category 7 alleles in Vista Bubblegum, and (10) all “out” alleles in the upright cultivar group B. The cultivars were arranged according to growth habit.
Genes involved in petunia axillary meristem outgrowth from previous reports were then placed within or adjacent to these genotype 7-only regions (Supplementary Table 3). Of these, we found that DAD1/CCD8 (AY746977.1) was located on chromosome 7 on scaffold Peaxi162Scf00227, which was near several transcripts harboring genotype 7-only alleles with the trailing cultivars. In addition, a P. ×hybrida TCP transcription factor 3 (TCP3) (KR002105.1) was found located on chromosome 3 and on the same scaffold that has transcripts within the outgroup allele introgression region, but both “out” alleles and the genotype 7 alleles were only present in ‘Wave Purple’ and ‘Surfinia Red’. For the genes with corresponding transcripts, the outgroup genotypes and the genotype 7 category distribution was further investigated. However, no transcripts had genotype 7 unique SNPs only present in the trailing cultivars (some of these were due to missing data in some of the cultivars).
For transcripts that had category 7 SNPs in every representative trailing cultivar, while no category 7 SNPs were present in any upright cultivars, gene ontology (GO) enrichment showed that the GO terms were enriched in the regulation of biological process and cellular process, many related to developmental process and response to stimulus (heat, chemical, light, etc.). For the molecular functions, we found a considerable number of transcription regulation and catalytic activity (Supplementary Table 4). These GO terms were similar with previously reported GO terms associated with growth habit in olive (Olea europaea L.)17. Longer internodes usually associated with trailing type while upright types had shorter internodes. In this set of genes, the carbohydrate metabolic processes and their regulation were enriched, consistent with the report on internode length in Lagerstroemia18. A total of 709 of these transcripts (unigenes) were annotated with the KO (Kyoto Encyclopedia of Genes and Genome (KEGG) Ortholog) database, and the most representative pathways were “Metabolic pathways” (98), “Biosynthesis of secondary metabolites” (44), and “RNA transport” (21) (Supplementary Table 5).
Discussion
The origin of P. ×hybrida was proposed to be from the breeding of only two species, one with white flowers (P. axillaris) and the other with purple flowers (P. integrifolia)16. Early petunia breeding was mainly through selection within the available hybrids3. However, by 1970 all cultivars produced a relatively uniform phenotype (large flowers on compact plants). Therefore, during the 1990s, petunia breeding programs started to introduce alleles from other Petunia species or old cultivars3. This process introduced a new phenotype—prostrate or trailing growth habit. The short yet complex breeding history shaped the current genome of petunia cultivars, which exhibit wide morphological variation19. Until now, the genetic background of commercial petunia cultivars remained uncharacterized. Elucidating the genetic diversity of petunia cultivars and inferring their genomic background from the progenitor species would facilitate the understanding of petunia breeding history and identification of the causal variants/genomic regions associated with important traits, such as stress tolerance and growth habit. In this study, transcriptome-derived SNPs were used to characterize the genetic variation among petunia cultivars.
P. integrifolia and P. axillaris were the hypothetical ancestors of petunia cultivars. P. exserta is not considered as a progenitor species2, but it may be useful for introgression of genes for novel traits such as flower color and shape, and growth habit, into the cultivated petunia3,20. P. integrifolia has five taxa and three of them could be a progenitor of the garden petunia3,4,21,22. P. exserta is the only ornithophilous species of the genus, with red flowers, protruded anthers, and stigma. Similar to previous findings that P. exserta and P. axillaris are closely related species22,23,24, in this study, >70% of the transcriptome-derived SNPs between the two species were monomorphic. The close phylogenetic relationship and the similar genetic distribution in PCA plots (Supplementary Fig. 1a) both support that the divergence between the two species is recent. On the genetic linkage map, we found regions harboring different genetic background between P. exserta and P. axillaris. These regions were detected on almost all linkage groups (chromosomes) and could contain important genes for habitat shift and floral traits or other factors involved in genetic isolation of these species. A number of recent reports and model predictions indicate the accumulation of differentiations/barrier genes among species in regions of low/restricted recombination25,26, including for Petunia27.
For the petunia cultivars, a range of genetic diversity was detected. On the PCA plot (Supplementary Fig. 1a), the three wild species were distributed toward the ends of both axes, while the cultivars tend to cluster in the middle. This could suggest that the majority of petunia cultivars behaved as true hybrids of the hypothetical progenitor species and had large genetic contribution from each of the two species.
Unlike many major crops, no signature of breeding was detected among petunia cultivars, possibly because of the short breeding history and the diversified breeding objectives. On the other hand, this could also be due to the decreased statistical power from the small sample size, even when potential candidate SNPs were identified by searching for SNPs with absolute alpha values (which indicate the strength and direction of selection) ranked within the top 5 and 10%.
We found that the P. ×hybrida genome contains genetic contribution from P. axillaris, P. integrifolia, and one or more unknown ancestors. This finding is consistent with previous research that showed both progenitor species and possibly other species contributed to the current genome of P. ×hybrida5. However, our study also showed that the proportion of contribution for each progenitor species varied across cultivars, though all cultivars contained a higher proportion of P. axillaris-unique alleles compared to P. integrifolia-unique alleles (Fig. 2). In addition, cultivars with similar proportions of P. axillaris and P. integrifolia contribution also had higher heterozygosity than those with a higher proportion of P. axillaris alleles. Heterozygosity levels for the traditional upright varieties averaged 10–15% (Fig. 1), which was surprisingly low considering that most are F1 hybrids. This suggests that the commercial P. ×hybrida germplasm pool may be of quite limited diversity. Identifying the genetic regions contributed by each of the progenitor species may provide a landscape of the recombination pattern across the petunia cultivar panel. However, at present, the genetic location on the linkage map does not directly correspond to the physical distance, as the recombination rate (cM/Mb) may vary on the genome and for each genotype.
In previous research on the P. ×hybrida genome, the majority of the genetic contribution was from P. axillaris5. A possible explanation for this difference could be the different representative P. ×hybrida genotypes used in the study. Commercial cultivars were used here, whereas three laboratory accessions were used formerly5. For the three laboratory lines, the cultivar ‘Mitchell’ was developed from a doubled haploid generated from a hybrid between P. axillaris and the backcross of the cultivar “Rose of Heaven” and P. axillaris F1 hybrid. Therefore, a high percentage of the P. axillaris genes was expected28. In the other two accessions, both had >10 generations of inbreeding, which could contribute to the genetic background bias toward one parent. We did not have enough resolution to further investigate the SNP distribution pattern at a gene level on the cultivar panel. Therefore, we could not verify on the petunia cultivars the previous findings that conserved blocks of SNPs similar to P. axillaris and other blocks similar to P. integrifolia between the previously reported genotypes could be due to gene conversion or random repair of heteroduplexes5.
The trailing cultivars have a short breeding history, dating back only to the 1990s3. The initial trailing petunias were reportedly developed by crossing P. ×hybrida with an unknown Petunia species29. Therefore, the prostrate or trailing phenotype was likely introduced through alleles of another wild species. In this study, SNPs of genotype category 7 [(P. axillaris = P. integrifolia)/out] were enriched in the representative trailing cultivars and thus added a piece of evidence that the trailing phenotype might be introduced from a third species. Considering all 14 petunia species are freely crossable with P. ×hybrida30, and P. altiplana has been reported to be “widely spreading and often forming a mat-like structure”31, unique within the genus2, we suspect that the trailing alleles in the cultivars were introgressed from P. altiplana.
However, if the trailing cultivars were the only type that had introgressed alleles from other species, the “out” alleles should be present only in these cultivars, which is inconsistent with the current findings. This could be due to: (1) the fact that exact progenitor genotypes for the petunia cultivars are unknown, therefore, some of the “true progenitors” alleles were not present in the current P. axillaris and P. integrifolia genotypes, which may have biased the genotype category estimation. It is also possible that the proportion of “out” alleles were overestimated; (2) the complex breeding history of petunia cultivars might have introduced alleles from other species than the two main progenitor species. Nevertheless, the most likely alleles for the trailing growth habit should be the “out” alleles that were only present in the trailing cultivars. Genome-wide comparisons of representative trailing cultivars with the compact cultivars showed that the introgressed “out” alleles were not restricted to a few discrete regions, instead, they were widespread and mostly heterogeneous loci across the genome.
Materials and methods
Transcriptome sequencing and read collection for petunia cultivars
A panel of 13 petunia cultivars with diverse growth habits was selected for transcriptome sequencing. Three of the cultivars—‘Wave Purple’ (trailing type), ‘Surfinia Red’ (trailing type), and ‘Madness Yellow’ (upright type), were selected to generate a reference transcriptome for P. ×hybrida. For these cultivars, tissues were collected from combined stages of floral development (from ca. 5 mm long buds through early fruit development) and leaves at different developmental stages (from ca. 1/10th up to 1/3rd of final expanded leaf length) pooled from five 10-week-old plants per cultivar and immediately frozen in liquid N2. For each of these cultivars, RNA extraction was carried out on pooled flower and leaf samples separately. To investigate the genomic background and genetic diversity of petunia cultivars, an additional ten cultivars were sequenced. For these ten cultivars, only leaf tissues were collected (same stages as above) and pooled for RNA extraction, again from five plants per cultivar. Of these cultivars, ‘Supertunia Yellow’, ‘Supertunia Vista Bubblegum (Vista Bubblegum)’, ‘Supertunia Vista Silverberry (Vista Silverberry)’, and ‘Tidal Wave Silver’ have a mounding/trailing habit; ‘Avalanche Salmon’ has a mounded habit; and ‘Fantasy Rose’, ‘Madness Red’, ‘Orchid Daddy’, ‘Storm Blue’, and ‘Super Cascade White’ have an upright habit (Table 1).
RNA was extracted using the Trizol® reagent (Life Technologies) following the manufacturer’s instructions. Forty micrograms of RNA was treated for DNA contamination with RNase-free DNase set (Qiagen). DNase-treated RNA was purified with the RNeasy® MinElute Cleanup Kit (Qiagen). RNA yield and quality were determined by Agilent 2100 BioAnalyzer RNA 6000 Pico chip (Agilent Technologies) with threshold RNA integrity number values ≥8.0. A TruSeq RNA Sample Preparation kit (Illumina) was used to construct the cDNA libraries. The six libraries of the leaf samples and flower samples for ‘Wave Purple’, ‘Surfinia Red’, and ‘Madness Yellow’ were pooled in one lane and sequenced on the Illumina HiSeq 2500 sequencer with 100 nt paired-end at the Michigan State University Research Technology Support Facility (RTSF; http://rtsf.msu.edu/; East Lansing, MI). For the other ten cultivars, the samples were barcoded and run on one lane of SE50 Illumina High Output flow cell at the Michigan State University RTSF. Sequences were filtered and trimmed with Trimmomatic with the corresponding functions for paired- and single-end reads, respectively32. Both raw and cleaned reads were assessed with FastQC v0.11.3 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
SNP detection, validation, and re-genotyping
Sequencing reads for the cultivar samples and wild species (P. axillaris, P. exserta, and P. integrifolia) reported previously6 were mapped to the P. axillaris transcriptome assembly using the Burrows–Wheeler Aligner program with default values for both single- and paired-end reads6,33. Duplicate reads were removed after the initial alignment to eliminate reads that mapped to the same position of the transcriptome. Duplicate removal was performed for the aligned reads using picardTools/1.89 (broadinstitute.github.io/picard). The subsequent local realignment to correct misalignments due to the presence of indels was performed using the Genome Analysis Toolkit (GATK)34. Initial variant calling was performed by HaplotypeCaller from GATK with a Phred-scaled confidence threshold of 30. After the initial variant calling, SNP filtering was performed using previously described criteria except with a modification to retain clustered SNPs (SNP clustering filter was removed) and with a minimum total depth of coverage of five for each genotype6. Afterwards, SNPs that showed no polymorphisms within the panel were excluded using the filters as previously described6.
SNP validation was carried out on 22 randomly chosen SNPs with no missing data on the genotype calling to ensure the maximum genotyping validation efficiency. The primers were designed around SNP loci where at least two species were polymorphic and on unigenes with exons >600 bp. Primer design, PCR conditions, and sequence visualization were as previously described6. The initial SNP validation identified that a small portion of the heterozygous genotypes were called as homozygous, therefore for the subsequent analyses, a series of custom genotyping filters were implemented to exclude low-quality reads or ambiguous genotypes and to re-define the genotype using allele read percentage. The filters were written by Python (Python 2.7.9)35 with the following criteria: (1) AA: SNPs were called homozygous and assigned to have the same genotype as the reference genotype (P. axillaris) when the reference reads were ≥80% of the total reads; (2) BB: SNPs were called homozygous alternative alleles if the alternative reads were >80% of the total reads; (3) AB: SNPs were called heterozygous if both the ratios of reference SNP reads to total reads at the loci and the ratio of the first alternative allele reads to entire reads at the loci were >0.3, and the sum of both ratios is ≥0.8. After the genotyping correction, SNPs were filtered to only keep the polymorphic loci.
Detecting signature of selection in petunia cultivars
To detect the presence of a breeding signature among the cultivars, the level of differentiation at a locus was compared with the level of differentiation across the transcriptome with 74,218 SNPs that had no missing data across the 16 genotypes. The FST outliers (loci deviating from neutral expectations) were detected by BayeScan v2.1 with default parameters15,36,37. BAYESCAN v. 2.163 implemented the Bayesian model decomposes FST values into locus-specific components (α) and population-specific components (β51). The method is based on a reversible jump MCMC algorithm and calculates the posterior probability of each under selection locus by assuming two alternative models, selection-based model and neutral model. A locus that is significantly more divergent than average is likely been affected by positive or direct selection. BayeScan can perform analyses on very small sample size without introducing bias15,36,37. The alpha values generated by BayeScan indicate the strength and direction of selection, with a positive value suggests diversifying selection and negative value suggests balancing or purifying selection38.
Dissecting the genetic background of petunia cultivars
Heterozygosity rate was calculated as the proportion of heterozygous SNPs for each individual (cultivars and the three wild species). Both the entire SNP dataset (1,164,566) and the set of SNPs with no missing data (74,218) were used for the heterozygosity rate estimation.
With the entire dataset (1,164,566 SNPs), two genotype classification approaches were used to understand the genetic background of petunia cultivars and their relationship with the ancestor species. The first approach (approach 1) followed the previous publication of Bombarely et al.5 with a slight modification. In brief, the cultivar genotypes were classified into 12 categories (Supplementary Table 1) by their nucleotide similarity with P. axillaris and P. integrifolia, where both P. axillaris and P. integrifolia were assumed to be homozygous and had different genotypes (0/0 for P. axillaris and 1/1 for P. integrifolia). Except that, at loci where P. axillaris and P. integrifolia had the same genotype calling, the cultivars with the same genotype as P. axillaris and P. integrifolia would be called homozygous P. axillaris = P. integrifolia, and any other alleles were categorized as from species other than P. axillaris and P. integrifolia (“out” alleles, when no polymorphisms were detected between P. axillaris and P. integrifolia, whereas the cultivar genotypes had a different allele at the locus). Unknown: Allele origin could not be determined due to the missing genotyping data in either P. axillaris or P. integrifolia. (Supplementary Table 1). SNPs with unknown genetic background due to missing data were assigned to the “unknown” category. The second approach (approach 2) did not directly estimate the cultivar genetic components, instead, a “thresholds model” was employed to assess the cultivar genotype proportion range for each category. One boundary assumed that the cultivar panels were derived from ancestral species that had exactly the same P. axillaris and P. integrifolia genotype as used in this study (where the P. integrifolia has >50% heterozygosity), therefore any cultivar genotypes that were the same as P. axillaris or P. integrifolia would be counted as derived from P. axillaris or P. integrifolia, and alleles not found in P. axillaris or P. integrifolia were assumed derived from other species (“out”). The second boundary assumed that both P. axillaris and P. integrifolia are homozygous, P. axillaris had the “0/0” genotype, and P. integrifolia had the “1/1” genotype. In such circumstances, the cultivars were dissected according to their similarity with “0/0” and “1/1”, and consequently the cultivars category classification was independent of the P. axillaris and P. integrifolia genotype calling.
Once the genetic background was identified for each cultivar, the percentage of homozygous P. axillaris SNPs for each transcript was projected to the genetic linkage map generated from an RIL population developed from the crossing of P. axillaris with P. exserta (AE map)11. The percentage of homozygous P. axillaris SNPs for a transcript was calculated by first assigning all SNPs for that cultivar on the transcript to a similarity rate based on their genotype categories (Supplementary Table 1). Subsequently, the average similarity was calculated for all SNPs on that transcript. Furthermore, a custom Python script was used to place the transcripts onto the AE map if a SNP on the map was located within the transcript covered range on the genome. Finally, the global percentages of SNP similarity with P. axillaris were calculated within each recombination interval. On the AE map, regions with low genetic recombinant frequency had only a few GBS-SNPs spanning long genetic distances. To avoid biased estimation, P. axillaris genetic similarity was not plotted on these regions. The percentage of SNP similarity with P. axillaris on the AE linkage groups was visualized using Circos39.
Exploring the genetic components difference for growth habit types in petunia cultivars
Two groups representing different growth habit types were used to study the genetic difference between the trailing and upright types. The trailing type group (Group A) had three cultivars: ‘Wave Purple’, ‘Surfinia Red’, and ‘Vista Bubblegum’; and the upright cultivar group (Group B) included ‘Orchid Daddy’, ‘Storm Blue’, and ‘Madness Yellow’. To inspect what genotype categories have been “enriched” in the trailing-type cultivars, the SNPs that were polymorphic between two groups were dissected into the 12 categories (approach 1), and the proportion of each category in the polymorphic SNP set and the original set were compared for each cultivar. In addition, the percentage of each category in the current polymorphic SNP set compared to the original SNP set were plotted for the cultivars, along with the average proportion of each category of polymorphic SNP in the entire SNP set plotted for comparisons.
After the candidate transcripts were identified (with alleles potentially introgressed from other species that might be related with growth habit), we performed a GO enrichment analysis using topGO package40 in R version 3.6 to identify if genes with particular molecular function and biological processes were overrepresented in these transcripts. The GO enrichment employed Fisher’s exact test for statistical significance. The enriched GO terms at p value of ≤0.05 were retained. Treemaps were produced to visualize enriched GO categories. Metabolic and the functional biological pathways were identified by mapping transcripts under the significantly enriched Go terms to the KEGG41 (http://www.genome.jp/kegg/) by KEGG Automatic Annotation Server42 (http://www.genome.jp/kegg/kaas/) with the available manually annotated plant databases and single-directional best hit.
Data availability
The raw data for RNA sequencing have been deposited in NCBI Sequence Read Archive (SRA) (SRA accession: PRJNA546569).
References
USDA-NASS. Floriculture crops 2018 summary. https://www.nass.usda.gov/Publications/Todays_Reports/reports/floran19.pdf (2019).
Stehmann, J. R., Lorenz-Lemke, A. P., Freitas, L. B. & Semir, J. in Petunia. Evolutionary, Developmental and Physiological Genetics (eds Gerats, T. & Strommer, J.) 1–28 (Springer, 2009).
Griesbach, R. J. in Flower Breeding and Genetics: Issues, Challenges and Opportunities for the 21st Century (ed. Anderson, N. O.) 301–336 (Springer, 2007).
Segatto, A. L. A., Ramos-Fregonezi, A. M. C., Bonatto, S. L. & Freitas, L. B. Molecular insights into the purple-flowered ancestor of garden petunias. Am. J. Bot. 101, 119–127 (2014).
Bombarely, A. et al. Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida. Nat. Plants 2, 16074 (2016).
Guo, Y., Wiegert-Rininger, K. E., Vallejo, V. A., Barry, C. S. & Warner, R. M. Transcriptome-enabled marker discovery and mapping of plastochron-related genes in Petunia spp. BMC Genomics 16, 726 (2015).
Broderick, S. R. et al. RNA-sequencing reveals early, dynamic transcriptome changes in the corollas of pollinated petunias. BMC Plant Biol. 14, 307 (2014).
Villarino, G. H., Bombarely, A., Giovannoni, J. J., Scanlon, M. J. & Mattson, N. S. Transcriptomic analysis of Petunia hybrida in response to salt stress using high throughput RNA sequencing. PLoS ONE 9, e94651 (2014).
Cao, Z. et al. Genome-wide Search for quantitative trait loci controlling important plant and flower traits in petunia using an interspecific recombinant inbred population of Petunia axillaris and Petunia exserta. G3 (Bethesda) 8, 2309–2317 (2018).
Cao, Z. et al. Genome-wide identification of quantitative trait loci for important plant and flower traits in petunia using a high-density linkage map and an interspecific recombinant inbred population derived from Petunia integrifolia and P. axillaris. Hortic. Res. https://doi.org/10.1038/s41438-018-0091-5 (2019).
Guo, Y., Lin, W. K., Chen, Q., Vallejo, V. A. & Warner, R. M. Genetic determinants of crop timing and quality traits in two interspecific Petunia recombinant inbred line populations. Sci. Rep. 7, 3200 (2017).
Chen, Q. C., Guo, Y. & Warner, R. M. Identification of quantitative trait loci for component traits of flowering capacity across temperature in Petunia. G3 (Bethesda) 9, 3601–3610 (2019).
Gerats, T. & Vandenbussche, M. A model system for comparative research: Petunia. Trends Plant Sci. Plant Model Syst. 10, 251–256 (2005).
Hoffmann, A. A. & Willi, Y. Detecting genetic responses to environmental change. Nat. Rev. Genet. 9, 421–432 (2008).
Foll, M. & Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180, 977–993 (2008).
Sink, K. C. in Monographs on Theoretical and Applied Genetics 9: Petunia (ed. Sink, K. C.) 3–9 (Springer, 1984).
González-Plaza, J. J. et al. Transcriptomic analysis using olive varieties and breeding progenies identifies candidate genes involved in plant architecture. Front. Plant Sci. 7, 240 (2016).
Ju, Y. et al. Transcriptome analysis of the genes regulating phytohormone and cellular patterning in Lagerstroemia plant architecture. Sci. Rep. 8, 15162 (2018).
Krahl, K. H. & Randle, W. M. Morphological variation in a petunia germplasm collection. Hort. Sci. 248, 482 (1993).
Warner, R. M. & Walworth, A. E. Quantitative inheritance of crop timing traits in interspecific hybrid Petunia populations and interactions with crop quality parameters. J. Hered. 101, 308–316 (2010).
Ando, T. et al. A morphological study of the Petunia integrifolia complex (Solanaceae). Ann. Bot. 96, 887–900 (2005).
Ando, T. et al. Phylogenetic analysis of Petunia sensu Jussieu (Solanaceae) using chloroplast DNA RFLP. Ann. Bot. 96, 289–297 (2005).
Kulcheski, F. R. et al. Molecular phylogenetic analysis of Petunia Juss. (Solanaceae). Genetica 126, 3–14 (2006).
Lorenz-Lemke, A. P. et al. Diversity and natural hybridization in a highly endemic species of Petunia (Solanaceae): a molecular and ecological analysis. Mol. Ecol. 15, 4487–4497 (2006).
Nachman, M. W. & Payseur, B. A. Recombination rate variation and speciation: theoretical predictions and empirical results from rabbits and mice. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 409–421 (2012).
Navarro, A. & Barton, N. H. Chromosomal speciation and molecular divergence–accelerated evolution in rearranged chromosomes. Science 300, 321–324 (2003).
Hermann, K. et al. Tight genetic linkage of prezygotic barrier loci creates a multifunctional speciation island in Petunia. Curr. Biol. 23, 873–877 (2013).
Mitchell, A. Z., Hanson, M. R., Skvirsky, R. C. & Ausubel, F. M. Anther culture of petunia: genotypes with high frequency of callus, root, or plantlet formation. Z. Pflanzenphysiol. 100, 131–145 (1980).
Rader, J. S. in Ball RedBook (ed. Ball, V.) (Ball Publishing, 1998).
Watanabe, H. et al. Cross compatibility of Petunia cultivars and P. axillaris with native taxa of Petunia in relation to their chromosome number. J. Jpn. Soc. Hort. Sci. 65, 625–634 (1996).
Ando, T. & Hashimoto, G. Two new species of Petunia (Solanaceae) from southern Brazil. Botanical J. Linn. Soc. 111, 265–280 (1993).
Bolger, A., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
van Rossum, G. & de Boer, J. Interactively testing remote servers using the Python programming language. CWI Q. 4, 283–303 (1991).
Foll, M., Fischer, M. C., Heckel, G. & Excoffier, L. Estimating population structure from AFLP amplification intensity. Mol. Ecol. 19, 4638–4647 (2010).
Fischer, M. C., Foll, M., Excoffier, L. & Heckel, G. Enhanced AFLP genome scans detect local adaptation in high-altitude populations of a small rodent (Microtus arvalis). Mol. Ecol. 20, 1450–1462 (2011).
Beaumont, M. A. & Balding, D. J. Identifying adaptive genetic divergence among populations from genome scans. Mol. Ecol. 13, 969–980 (2004).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Alexa, A. & Rahnenführer, J. Gene set enrichment analysis with topGO. https://bioconductor.org/packages/release/bioc/vignettes/topGO/inst/doc/topGO.pdf (2009).
Kanehisa, M. KEGG bioinformatics resource for plant genomics and metabolomics. Methods Mol. Biol. 1374, 55–70 (2016).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185 (2007).
Acknowledgements
Funding was provided by USDA Specialty Crops Research Initiative Award number 2011-51181-30666 to R.M.W. R.M.W. is supported in part by Michigan AgBioResearch and through USDA National Institute of Food and Agriculture, Hatch project number MICL02451.
Author information
Authors and Affiliations
Contributions
Y.G. and R.M.W. designed the experiments and wrote the manuscript. Y.G. generated and analyzed the data. Both authors read and approved the submitted version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, Y., Warner, R.M. Dissecting genetic diversity and genomic background of Petunia cultivars with contrasting growth habits. Hortic Res 7, 155 (2020). https://doi.org/10.1038/s41438-020-00373-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-020-00373-2
