
Abstract
Lauraceae includes the genus Phoebe, and the family is linked to the evolution of magnoliids. We sequenced the genome of Phoebe bournei Nanmu. The assembled genome size was 989.19 Mb, with a contig N50 value of 2.05 Mb. A total of 28,198 protein-coding genes were annotated in P. bournei. Whole-genome duplication (WGD) analysis showed that Lauraceae has experienced two WGD events; the older WGD event occurred just before the divergence of Lauraceae and Magnoliales, and the more recent WGD was shared by all lineages of Lauraceae. The phylogenetic tree showed that magnoliids form a sister clade to monocots and eudicots. We also identified 63 MADS-box genes, including AGL12-like genes that may be related to the regulation of P. bournei roots and FIN219-like genes encoding GH3 proteins, which are involved in photomorphogenesis. SAUR50-like genes involved in light signal-mediated pedicel or stem development were also identified. Four ATMYB46- and three PtrEPSP-homologous genes related to lignin biosynthesis were identified. These genes may be associated with the formation of straight trunks in P. bournei. Overall, the P. bournei reference genome provides insight into the origin, evolution, and diversification of Phoebe and other magnoliids.
Similar content being viewed by others


Introduction
Lauraceae belongs to Laurales, which together with Canellales, Piperales, and Magnoliales, constitute the magnoliids, including 9000 species1,2,3. The relationships among the magnoliids, eudicots, and monocots remain unclear, even after the publication of four magnoliid genomes4,5,6,7,8. The genomes of Piper nigrum, Persea americana, and Liriodendron chinense support the magnoliids as a sister clade of monocots and eudicots4,5,6, while the genome of stout camphor Cinnamomum kanehirae supports magnoliids as a sister clade of eudicots7. The conflicts in terms of phylogenetic tree location indicate that additional genomic data are needed to more clearly elucidate the relationships among magnoliids, eudicots, and monocots, especially the species within Lauraceae.
Lauraceae is an important economic and ecological family including 2850 species of herbs, shrubs, and trees, mainly distributed in tropical and subtropical regions of Asia and South America9. The genus Phoebe within Lauraceae includes ~100 species of evergreen trees and shrubs10. Phoebe bournei (Nanmu) is endemic to China, where it is a protected species10. Wood from P. bournei, known as “wood with golden wire”, is used in the production of high-quality furniture and handicrafts due to its vertical wood texture, unique fragrance, resistance to insects and rot, durability, and beauty11,12. In ancient times, P. bournei timber was often used in palace construction to produce columns that represented the power and status of the nobles10,13,14. P. bournei is often used as a street tree because of its straight trunk and broad crown15. Due to intensive deforestation, poor seed germination, slow growth, and illegal timber logging, natural populations of P. bournei are now fragmented and threatened11,12,13,14,15,16,17.
Here, we report a reference genome of P. bournei obtained using the PacBio sequencing platform. The results can help to reveal its phylogenetic position within the magnoliids. Analysis of the P. bournei genome will provide insights into the demographic history of magnoliids, and data for future conservation efforts and biological research.
Results and discussion
Genome sequencing and assembly
To completely sequence the P. bournei genome, a total of 102.05 Gb of raw data were generated from 500 bp-insert libraries by Illumina sequencing (Supplementary Table 1). Survey analysis indicated that the P. bournei genome shows a high level of heterozygosity, corresponding to 1.54% of the 1.00 Gb genome sized according to 19 K-mer analysis (Supplementary Fig. 1). For the de novo whole-genome sequencing of P. bournei, we obtained 109.83 Gb of raw data using PacBio sequencing (Supplementary Fig. 2 and Supplementary Table 2). The assembled genome was 989.19 Mb, with a contig N50 value of 2.05 Mb (Supplementary Table 3). Benchmarking Universal Single-Copy Orthologs (BUSCO)18 assessment showed that the completeness of the gene set of the assembled genome was 95% (Supplementary Table 3), and the Illumina read alignment rate was 98.87% (Supplementary Table 4), indicating that the P. bournei genome assembly was of high quality and could be used for subsequent analysis.
Gene prediction and annotation
We annotated 28,198 protein-coding genes from the assembled P. bournei genome, 95.44% of which were supported by de novo and transcriptome data (Supplementary Fig. 3 and Supplementary Table 5). The proteome of the protein-coding genes of P. bournei was estimated to be 81.1% complete based on BUSCO analysis (Supplementary Table 6)18. We also identified 145 microRNAs, 813 transfer RNAs, 2417 ribosomal RNAs, and 519 small nuclear RNAs (Supplementary Table 7).
Through a combination of homology-based searches and de novo prediction, we estimated that 68.51% of the P. bournei genome consisted of repetitive sequences, with LTR/Gypsy sequences accounting for 25.22% of the genome and LTR/Copia sequences accounting for 13.67% (Supplementary Figs. 4, 5 and Supplementary Tables 8, 9). The C. kanehirae genome showed 20.5% fewer repeat sequences than the P. bournei genome, while the abundance of LTR/Gypsy and LTR/Copia sequences was 15.53% and 10.21% greater, respectively, than in P. bournei7. The insertion time of the LTR, Copia, and Gypsy elements in P. bournei was ~0.2 million years ago (Supplementary Fig. 6). We identified 27,011 genes that were annotated in seven functional databases. Among these genes, 19,417 (68.86%) were annotated with KOG terms, 19,649 (69.68%) were annotated with KEGG Ortholog terms, and 15,470 (54.86%) were annotated with Gene Ontology terms (Supplementary Table 10).
Evolution of gene families
Laurales belongs to the magnoliids, and the evolutionary position of magnoliids is still unclear4,5,6,7,8,19,20. We constructed a high-confidence phylogenetic tree based on 292 single-copy gene families extracted from the genomes of 18 species (Supplementary Fig. 7 and Supplementary Table 11). The phylogenetic trees were constructed using the phase 1 loci of orthologous genes, with the branch lengths representing evolutionary rates. The phylogenetic tree showed that magnoliids, including C. kanehirae, P. bournei, Persea americana, Litsea cubeba, P. nigrum, and L. chinense, formed a sister group to the monocot−eudicot clade (Fig. 1a) based on the Bayesian method. This is consistent with the phylogenetic trees of magnoliids based on the L. chinense5 genome and the P. americana6 genome. To further determine the positional relationships of magnoliids, monocots, and eudicots, we constructed concatenated and ASTRAL trees based on protein and nucleotide sequences. The phylogenetic trees constructed via the ASTRAL and concatenated methods based on amino acids also showed that the magnoliid clade was sister to the monocot-eudicot clade (Fig. 1b; Supplementary Fig. 8). However, the phylogenetic trees constructed via the ASTRAL and concatenated methods based on nucleotides provided support for a recent common ancestor of magnoliids and monocots, which formed a clade sister to the eudicot clade (Fig. 1c). Incomplete lineage sorting is better understood as a problem of ancestral polymorphism that does not sort according to the species tree, and this polymorphism is represented in terms of haplotypes or alleles. Copy number variations can also help define haplotypic or allelic states, and copy number variants (e.g., tandem duplicates) can contribute to incomplete lineage sorting. Thus, genealogies based on orthology remain difficult to differentiate from those based on orthology plus paralogy. Therefore, we favored a phylogenetic tree based on the Bayesian method. In addition, using the MCMC tree with fossil calibration, we estimated that the Lauraceae divergence time was 127.56 Mya, and the divergence time between P. bournei and P. americana was 14.05 Mya (Supplementary Fig. 9).

a Phylogenetic tree based on the Bayesian method. b Phylogenetic tree based on the ASTRAL (left) and concatenated (right) amino acid sequences. c Phylogenetic tree based on the ASTRAL (left) and concatenated (right) nucleotide sequences
The expansion and contraction of orthologous gene families were determined based on a probabilistic graphical model (Fig. 2). A total of 1075 gene families were expanded in the lineage leading to Laurales, and 547 families were contracted. A total of 745 gene families were expanded in P. bournei, compared with 1198 and 910 in P. americana and C. kanehirae, respectively. At the same time, 1785 gene families were contracted in P. bournei, compared with 1626 and 1044 in P. americana and C. kanehirae, respectively.

The green numbers are the numbers of expanded gene families, and the red numbers are the numbers of contracted gene families. The blue portions of the pie charts represent the gene families whose copy numbers are constant. The orange portions of the pie charts represent the proportion of 11,968 gene families found in the most recent common ancestor (MRCA) that have expanded or contracted during recent differentiation
Synteny analysis and whole-genome duplications (WGD)
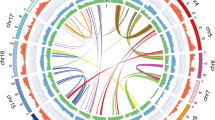
The distribution of Ks values in the P. bournei, C. kanehirae, P. americana, and L. cubeba genomes showed two clear peaks, one at Ks1 ≈ 0.5–0.6 and the other at Ks2 ≈ 0.85–0.95 (Fig. 3a). These two Ks peaks were greater than the Ks values of the differentiation peaks of P. bournei–C. kanehirae, P. bournei–L. cubeba, and P. bournei–P. americana (Ks < 0.1). This result indicated that the common ancestor of Lauraceae (P. bournei, C. kanehirae, L. cubeba, and P. americana) underwent two polyploidization events before the groups diverged. Collinearity analysis confirmed that these two polyploidy events of P. bournei were WGD events (Fig. 3b). C. kanehirae, L. cubeba, and P. americana also experienced two WGD events6,7,8. The distribution of Ks values in the P. nigrum genome showed one peak, which means that one WGD event occurred in the P. nigrumi genome. This is consistent with previously reported WGD results for the P. nigrumi genome4. The Ks differentiation peak of P. bournei–P. nigrum occurred at Ks ≈ 1.75, which was greater than the two Ks peaks (Ks1 ≈ 0.5–0.6 and Ks2 ≈ 0.85–0.95) observed in Lauraceae (P. bournei, C. kanehirae, L. cubeba, and P. americana) genomes (Fig. 3a). This result indicates that after the divergence of the common ancestors of Lauraceae and Piperales (P. nigrum), Lauraceae experienced two WGD events. The Ks differentiation peak of P. bournei–L. chinense (Ks ≈ 0.825) was larger than the Ks1 peak (Ks1 ≈ 0.5–0.6) in Lauraceae (P. bournei, C. kanehirae, L. cubeba, and P. americana) and smaller than the Ks2 peak (Ks2 ≈ 0.85–0.95) in Lauraceae (P. bournei, C. kanehirae, L. cubeba, and P. americana). The results showed that an ancient WGD event (Ks2 ≈ 0.85–0.95) occurred in Lauraceae genomes before the differentiation of L. chinense (Magnoliales) and Lauraceae and that a recent WGD event (Ks1 ≈ 0.5–0.6) occurred after the differentiation of L. chinense (Magnoliales) and Lauraceae. The gene tree and Ks analysis both showed that L. chinense experienced one WGD event and revealed two WGDs in the P. bournei genome (Fig. 3a; Supplementary Fig. 10). Based on the previous WGD analysis of the L. cubeba genome8, we suggest that Lauraceae has experienced two WGD events: an ancient WGD event, which may have occurred just before the divergence of Magnoliales and Lauraceae, and a recent WGD, occurring before the differentiation of Lauraceae (Fig. 3c).

a Ks distribution in P. bournei and other species. Peaks in the intraspecies Ks distribution indicate whole-genome polyploidization events, and peaks in the interspecies Ks distribution indicate speciation events. b Collinear point diagram of the P. bournei genome. There are multiple 1:3 collinear regions in the P. bournei genome, among which one extra green copy region provides evidence of recent WGD, and two extra red copy regions provide evidence of ancient WGD events. c The phylogeny of magnoliids with WGD events
MADS-box gene family analysis
The MADS-box gene family participates in many plant processes, including floral development, flowering time determination, and fruit ripening21. A total of 63 MADS-box genes were identified in the P. bournei genome, which were classified into type I and type II MADS-box genes based on phylogenetic analysis. Thirty type I MADS-box genes were subdivided into three subfamilies: Mα, Mβ, and Mγ (Table 1; Supplementary Table 12). There were three and four members in Mγ and Mβ, respectively. The orthologs of Mα have been duplicated (23 members). Type I genes have been associated with the development of the female gametophyte, embryo22, and central cell and endosperm23,24. Their specific roles in P. bournei are unknown.
Type II MADS-box genes were divided into 27 MIKCC-type and six MIKC*-type MADS-box genes (Fig. 4; Supplementary Table 12). MIKC* regulation affects pollen gene expression25,26. There were fewer genes from the SOC1-class (three members), A class (two members), and AGL6 clade (one member) than in A. thaliana. The AGL12 and ANR1 genes are involved in root development27,28. P. bournei and A. thaliana both contain four ANR1 clade genes. P. bournei exhibits more AGL12 genes (four members) than A. thaliana. P. bournei exhibits more genes related to root development, possibly because it requires strong roots to support its growth. However, we did not observe FLC subfamily genes, indicating that this family may be absent in P. bournei, possibly because P. bournei does not require vernalization for flowering, similar to rice29. Bs-class genes are usually involved in seed pigmentation and endothelium development30. However, there are no genes related to the Bs class in P. bournei. This difference might be related to the different fruit- and seed-development processes among species.

The gene ID numbers begin with “Pb” to represent the gene IDs of P. bournei, “Ck” to represent the gene IDs of C. kanehirae, and “AT” to represent the gene IDs of A. thaliana
GH3 and SAUR gene family analysis
Auxin regulates plant growth and development by altering the expression of multiple genes31. Auxin-responsive genes can be divided into three classes: auxin/indoleacetic acid protein (Aux/IAA), small auxin-up RNA (SAUR), and glycoside hydrolase 3 (GH3) genes32. The GH3 gene family belongs to the auxin-responsive gene family. GH3 genes encode a class of acylamide synthetases that bind amino acids to indole-3-acetic acid (IAA), jasmonic acid (JA), and salicylic acid (SA). This changes the concentration of their bioactive forms in cells and regulates plant growth, development, and defense responses33,34,35. GH3 proteins are classified into three groups: group I, with JA and/or SA-amido synthetase activity; group II, with IAA–amido synthetase activity; and group III, with unknown synthetase activity36,37. We identified 17 and 14 GH3-class genes in P. bournei and C. kanehirae, respectively, and divided them into the three groups based on phylogenetic information (Fig. 5a). There were eight genes in groups I and II in P. bournei. There were seven genes in group I, and six genes in group II in C. kanehirae. P. bournei and C. kanehirae each exhibited one gene in group III.

a Phylogenetic relationships of P. bournei, C. kanehirae, and A. thaliana GH3 proteins. Bootstrap values are indicated at each node. The gene ID numbers begin with “Pb“ to represent the gene IDs of P. bournei, “Ck” to represent the gene IDs of C. kanehirae, and “AT” to represent the gene IDs of A. thaliana. In the parentheses, the number on the left is the number of GH3 homologs in the genome of P. bournei, and that on the right is the number of GH3 homologs in the genome of C. kanehirae. b Expression patterns of GH3 in the cortex, heartwood, and leaf. “Co” represents the cortex, “He” represents heartwood. “Le” represents the leaf. The accession numbers of the GH3 genes of A. thaliana are shown in Supplementary Table 13
ATGH3.5, ATGH3.6, and ATGH3.1 encode IAA–amido synthetases, which help to maintain auxin homeostasis by conjugating IAA to amino acids38. The orthologous genes (Pb42207 and Pb52834) of ATGH3.5, ATGH3.6, and ATGH3.1 are expressed in the heartwood and cortex (Fig. 5b). We also found one ATGH3.9-orthologous gene, Pb48914, showing high expression in the heartwood and cortex (Fig. 5b). ATGH3.9 controls auxin activity through amino acid conjugation and promotes primary root growth39. The ATGH3.9-orthologous gene in P. bournei shows the same function in the heartwood and cortex, thus promoting stem growth. In addition, FIN219 (ATGH3.11), a phytochrome A signaling component, plays a crucial role in photomorphogenesis40. We identified five FIN219-orthologous genes in P. bournei and four FIN219-orthologous genes in C. kanehirae (Fig. 5a). Expression analysis showed that three FIN219-orthologous genes (Pb06967, Pb41896, and Pb16041) in P. bournei were expressed in the heartwood, cortex, and leaves, while one FIN219-orthologous (Pb16231) gene was expressed only in the heartwood and leaves (Fig. 5b).
The SAUR gene family encodes highly unstable mRNA molecules with a very high turnover rate that are induced within minutes after auxin application. SAUR proteins promote cell elongation41. We identified 77 SAUR gene family members from P. bournei and 76 SAUR gene family members from C. kanehirae (Fig. 6). The Arabidopsis AtSAUR50 gene is involved in light signal-mediated pedicel development42. In sunflower (Helianthus annuus), the SAUR50-like gene is more highly expressed on the eastern side of stems during the day, leading to sun-tracking movement during the seedling stage43. We also identified two SAUR50-like genes in P. bournei (Pb20906 and Pb54747) and C. kanehirae (Fig. 6). FIN219-like genes and SAUR50-like genes in P. bournei may enable P. bournei to intercept more sunlight in dense forests, thus straightening the trunk.

The gene ID numbers begin with “Pb” to represent the gene IDs of P. bournei, “Ck” to represent the gene IDs of C. kanehirae, and the “AT” to represent the gene IDs of A. thaliana. SAUR50-like genes of P. bournei are indicated in red
Lignin-formation analysis
The principal components of wood are lignocellulosic polymers, which represent the most abundant biomass produced by terrestrial plants. Lignin biosynthesis evolved from the phenylpropanoid pathway, which promoted the successful colonization of terrestrial environments by plants44,45. The R2R3-MYB gene family regulates specific processes in plants, including phenylpropanoid biosynthesis46. MYB46-class genes mainly regulate the phenyl propyl pathway and lignin biosynthesis in plants. A. thaliana MYB46 directly targets and activates the expression of multiple lignin biosynthetic genes, such as MYB58 and MYB6347. There are four homologs of MYB46 in Populus trichocarpa (PtrMYB002, PtrMYB003, PtrMYB020, and PtrMYB021), and they all activate the promoters of lignin biosynthetic genes46,48,49. Our phylogenetic tree showed that four MYB46-homologous genes and six MYB46-homologous genes were present in the P. bournei and C. kanehirae genomes, respectively (Fig. 7a; Supplementary Fig. 11). All MYB46 orthologous genes from P. bournei were expressed in the heartwood and at low levels in the cortex, but were not in the leaves (Fig. 7c). Xie et al.50 found that a 5-enolpyruvylshikimate 3-phosphate synthase gene of P. trichocarpa (PtrEPSP) can directly bind to the promoter and repress the expression of a SLEEPER-like transcriptional regulator, which itself specifically binds to the promoter and represses the expression of PtrMYB021, thereby affecting lignin biosynthesis. We identified three PtrEPSP-homologous genes in the P. bournei and C. kanehirae genomes (Fig. 7b). PbEPSP1 was highly expressed in the heartwood, cortex, and leaves, but PbEPSP3 was not expressed (Fig. 7c).

a Phylogenetic relationship of PbMYB46-homologous genes with anther secondary wall- or lignin-associated MYBs. b Phylogenetic relationships of PbEPSP-homologous genes with EPSP-homologous genes in different plants. c Expression patterns of MYB46 and EPSP in the cortex, heartwood, and leaves. “He” represents heartwood, “Co” represents the cortex, and “Le” represents leaves. The accession numbers of MYB46 and EPSP in different species are shown in Supplementary Table 13
Conclusion
We assembled 989.19 Mb of the P. bournei genome and annotated 28,198 protein-coding genes. Although the genomes of four species within the magnoliid family have been sequenced, the phylogenetic relationships of the magnoliids are unclear. Our study showed that magnoliids are a sister clade to monocots and eudicots. WGD analysis suggested that Piperales and Magnoliales have both experienced one WGD event, and that Lauraceae has experienced two WGD events, where the older WGD event is shared with Magnoliales and the younger with other Lauraceae species. We identified 63 MADS-box genes in P. bournei and four AGL12-like and four ANR1-like genes may be related to the regulation of the roots. GH3 proteins are involved in regulating plant growth, development, and defense responses, and SAUR proteins promote cell elongation. We identified 17 members of the GH3 gene class and 77 members of the SAUR gene class in P. bournei. Five FIN219-like genes involved in photomorphogenesis and two SAUR50-like genes involved in light signal-mediated pedicel or stem development were identified. MYB46 and PtrEPSP activate the promoters of lignin biosynthetic genes, and we identified four homologous genes of MYB46 and three homologous genes of PtrEPSP in the P. bournei genome. These genes may be related to the formation of straight trunks in P. bournei. The P. bournei genome provides new insight into magnoliid genome evolution and diversification.
Materials and methods
DNA preparation and sequencing
All of the plant materials used in this study were collected from a mature P. bournei tree growing in Fujian Agriculture and Forestry University, Fujian Province, China. Total genomic DNA was extracted with a modified cetyltrimethylammonium bromide (CTAB) method for Illumina and de novo sequencing and assembly. Five-hundred bp paired-end libraries were constructed using the Illumina protocol. Genome size and heterozygosity were measured using GenomeScope51 based on a 19 K-mer distribution. In addition, we constructed SMRT libraries using the PacBio 20-kb protocol (https://www.pacb.com/), and they were subsequently sequenced on the PacBio platform. The transcriptomes of the heartwood, cortex, and leaves were sequenced on the Illumina platform.
Genome assembly
Canu52 was used to correct errors in the original data. Flye v2.4.253 was used to assemble the corrected data. Because of the high error rate of the de novo data, indel and SNP errors still existed in the assembly results. Thus, Arrow (https://github.com/PacificBiosciences/GenomicConsensus) was used to correct the assembly results. We compared the second-generation small fragment data with the assembly results, and further corrected the assembly results with Pilon v1.2254 to eliminate indel and SNP errors. The assembled sequence was larger than the genome size estimated through K-mer analysis, so we used trimDup (Rabbit Genome Assembler: https://github.com/gigascience/rabbit-genome-assembler) to remove redundancy from the assembly results. To confirm the quality of the genome assembly, we performed a BUSCO v4 (https://busco.ezlab.org)18 assessment using single-copy orthologous genes.
Identification of repetitive sequences
Repetitive sequence annotation was mainly based on homologous sequence alignment and de novo assembly. Homologous sequence alignment was based on the RepBase v21.12 database55 (http://www.girinst.org/repbase), and RepeatMaske v4.0.756 and RepeatProteinMask v4.0.756 were used to identify sequences similar to known repeat sequences. We identified TEs in the P. bournei genome using RepeatModeler (http://www.repeatmasker.org/RepeatModeler/)57 and LTR_FINDER v1.06 (http://tlife.fudan.edu.cn/ltr_finder/)58. In addition, tandem repeats across the genome were predicted using Tandem Repeats Finder v4.0959 (http://tandem.bu.edu/trf/trf.html). Finally, repeat sequences with identities ≥50% were grouped into the same classes.
Gene prediction and annotation
Two independent methods were used to predict protein-coding genes: homology-based and de novo-based prediction. Homologous proteins from nine known whole-genome sequences of Amborella trichopoda, Aquilegia coerulea, A. thaliana, C. kanehirae, Ginkgo biloba, L. chinense, Picea abies, P. trichocarpa, and Vitis vinifera were aligned to the P. bournei genome sequence using Exonerate v2.2.0 (https://www.ebi.ac.uk/Tools/psa/genewise/)60 for homology-based prediction. The sequences of these known genomes were downloaded from Phytozome 12 (https://phytozome.jgi.doe.gov/pz/portal.html). Two ab initio prediction software programs, Augustus61 (http://bioinf.uni-greifswald.de/augustus/) and SNAP62 (http://homepage.mac.com/iankorf), were used for de novo gene prediction. Then, the homology-based and ab initio gene structures were merged into a nonredundant gene model using Maker63 (http://weatherby.genetics.utah.edu/MAKER/wiki/index.php/MAKER_Tutorial_for_WGS_Assembly_and_Annotation_Winter_School_2018). We further filtered the annotated results of Maker, with the following genes filtered: (1) protein length < 50 aa and homologous protein support for exon region < 50%; and (2) CDS of the coding region and TE overlap length > 80%.
To obtain gene function information, we used BLAST v2.2.3164 to align the annotation results with seven protein databases, including SwissProt (http://www.uniprot.org)65, TrEMBL (http://www.uniprot.org/)65, KEGG (http://www.genome.jp/kegg/)66, InterPro (https://www.ebi.ac.uk/interpro/)67, NR, KOG68, and GO69. The tRNAs were predicted using tRNAscan-SE 1.3.170. The rRNAs were identified by aligning the rRNA template sequences from the Rfam database71 against the genome using the BLASTN algorithm. The miRNAs and snRNAs were predicted using INFERNAL (http://infernal.janelia.org/)72 in Rfam, and other ncRNAs were predicted with Infernal software (http://infernal.janelia.org/)72 against the Rfam database.
Genome-evolution analysis
Genes from the whole-genome sequences of 18 species (P. bournei, Ananas comosus, A. thaliana, A. trichopoda, Phalaenopsis equestris, P. trichocarpa, Solanum lycopersicum, Spirodela polyrhiza, V. vinifera, L. chinense, Oryza sativa, L. cubeba, C. kanehirae, P. americana, Asparagus officinalis, Actinidia chinensis, Nymphaea colorata, and P. nigrumi) were used for gene family-clustering analysis. We first constructed the protein data sets of these genomes and then used BLASTP (E-value of 1E-5) to align the protein data sets with themselves and to filter out low-quality sequences53. Orthologous groups present in the 18 genomes were identified using OrthoMCL v1.4 (http://orthomcl.org/orthomcl/)73.
MUSCLE (http://www.drive5.com/muscle/)74 was used to align the amino acid sequences of single-copy orthologous groups. The nucleotide sequences of the single-copy orthologous groups were connected into a supergene, and the data set was employed to construct a phylogenetic tree by using the GTR + gamma model in Mrbayes75. In addition, we used RAxML to combine all the data sets and constructed phylogenetic trees for protein and CDS sequences via concatenation and ASTRAL methods, respectively.
The data set employed for phylogenetic analysis was used to estimate the divergence times of each tree node using the MCMCTREE program (http://abacus.gene.ucl.ac.uk/software/paml.html) of the PAML package v4.776. The nucleic acid replacement model was the GTR model, and the molecular clock model was the independent rate model. The MCMC process consisted of 500,000 burn-in iterations and 500,0000 sampling iterations (sampling every 100 iterations). The same parameters were executed twice to obtain a more stable result. Published data from Lemna minor–O. sativa (117–140 Mya), magnolias (112.6 Mya), monocots–dicots (140 Mya), and angiosperms (200 Mya) were used to calibrate divergence times77,78.
We used CAFÉ 4.2 software (http://sourceforge.net/projects/cafehahnlab/)79 to measure the expansion and contraction of orthologous gene families. Based on the maximum-likelihood modeling of gene gains and losses, we analyzed gene families for signs of expansion or contraction using genome data from 18 species.
Collinearity analysis and whole-genome duplication
Within collinear segments, genes are conserved in function and sequence, and these genes remain highly conserved during the evolution of species. We used the default parameters of JCVI v0.9.14 (https://pypi.org/project/jcvi/)80 to analyze the protein sequences of P. bournei, P. americana, C. kanehirae, L. cubeba, L. chinense, and P. nigrum, and obtained the gene pairs in the collinear regions. We used Ks distribution analysis to estimate WGD events in the P. bournei, P. americana, C. kanehirae, L. cubeba, L. chinense, and P. nigrum genomes. Diamond was used to conduct self-alignment of the protein sequences of these species genomes and then to extract the mutual optimal alignment in the alignment results. Finally, Codeml in the PAML package was used to calculate Ks values81,82.
Our Ks analysis showed that the genomes of P. bournei presented two Ks peaks (Ks1 ≈ 0.5–0.6 and Ks2 ≈ 0.85–0.95), whereas that of L. chinense only exhibited one (Ks ≈ 0.7), and P. bournei–L. chinense showed one differentiation peak (Ks ≈ 0.825) (Fig. 3). Therefore, to determine the differentiation of P. bournei and L. chinense, we constructed a gene tree. For the construction of the gene tree, Blastp (e-value < 1e-5)53 was first used to align the protein sequences of all pairs of genes in the P. bournei genome equal to the Ks peak of P. bournei itself with those in the L. chinense genome equal to the Ks peak of L. chinense itself. Then, according to the alignment result, the genes meeting one of the following two conditions were selected: gene pairs of the Ks2 peak in the P. bournei genome were aligned to the gene pairs of the Ks peak in the L. chinense genome; the gene pairs of the Ks1 and Ks2 peaks in the P. bournei genome were aligned to the gene pairs of the Ks peak in the L. chinense genome. We selected nine gene pairs that satisfied the first condition and two gene pairs that satisfied the second condition. Finally, the selected gene pairs were employed to construct a gene tree using RaxMLv8 (parameter -m PROTGAMMAJTT), and the outgroup was fixed as evm_27.model.amtr_v1.0_scaffold00106.118 of A. trichopoda83.
Gene family analysis
The candidate sequences of the MADS-box and SAUR genes of A. thaliana were downloaded from TAIR (https://www.arabidopsis.org/index.jsp). The HMM profiles of the MADS (PF00319)84, GH3 auxin-responsive promoter (PF03321)85, and SAUR (PF02519)86 gene families were obtained from Pfam (http://pfam.xfam.org/). Each protein of the gene families of P. bournei and C. kanehirae was separately searched with the HMMER 3.2.1 (with default parameters)87 and BLASTP (E-value of e−5)53 methods in the P. bournei and C. kanehirae genomes. The whole-genome sequence of C. kanehirae was extracted from NCBI (Bioproject: PRJNA477266). The protein sequence set for the MADS-box gene candidates of P. bournei was employed for BLAST analysis against the assembled P. bournei transcriptomes with the TBLASTN program53. Subsequently, all of the candidate sequences of the MADS-box, GH3, and SAUR genes were subjected to SMART analysis (http://smart.embl-heidelberg.de/)88. The candidate MADS-box, GH3, and SAUR gene families were aligned with MEGA589, and the phylogenetic tree was constructed on the CIPRES website (https://www.phylo.org/portal2/). The MADS-box, CH3, and SAUR phylogenetic trees were visualized using iTOL (https://itol.embl.de).
Data availability
Genome sequences have been submitted to the National Genomics Data Center (NGDC). The raw whole-genome data of P. bournei have been deposited in BioProject/GSA (https://bigd.big.ac.cn/gsa.)90 under the accession codes PRJCA002001/CRA002192, and the assembly and annotation of the whole-genome data have been deposited at BioProject/GWH (https://bigd.big.ac.cn/gwh)91 under the accession codes PRJCA002001/GWHACDM00000000.
References
Zeng, L. et al. Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times. Nat. Commun. 5, 4956 (2014).
Sauquet, H. & Magallón, S. Key questions and challenges in angiosperm macroevolution. N. Phytol. 219, 1170–1187 (2018).
Chase, M. et al. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20 (2016).
Hu, L. et al. The chromosome-scale reference genome of black pepper provides insight into piperine biosynthesis. Nat. Commun. 10, 1–11 (2019).
Chen, J. F. et al. Liriodendron genome sheds light on angiosperm phylogeny and species–pair differentiation. Nat. Plants 5, 18–25 (2019).
Martha, R. A. et al. The avocado genome informs deep angiosperm phylogeny, highlights introgressive hybridization, and reveals pathogen-influenced gene space adaptation. Proc. Natl Acad. Sci. USA 116, 17081–17089 (2019).
Chaw, S. M. et al. Stout camphor tree genome fills gaps in understanding of flowering plant genome evolution. Nat. Plant 5, 63–73 (2019).
Chen, Y. C. et al. The Litsea genome and the evolution of the laurel family. Nat. Commun. 11, 1675 (2020).
Christenhusz, M. J. M. & Byng, J. W. The number of known plants species in the world and its annual increase. Phytotaxa 261, 201–217 (2016).
Li, Y. et al. Complete chloroplast genome sequences of two endangered Phoebe (Lauraceae) species. Bot. Stud. 58, 37–46 (2017).
Ding, X. et al. Congruent species delimitation of two controversial gold‐thread P. bournei tree species based on morphological and restriction site‐associated DNA sequencing data. J. Sys Evol. 57, 234–246 (2019).
Ge, Y. J. et al. Fengshui forests conserve genetic diversity: a case study of Phoebe bournei (Hemsl.) Yang in southern China. Genet. Mol. Res. 14, 1986–1993 (2015).
Ding, Y. et al. Development of EST-SSR markers and analysis of genetic diversity in natural populations of endemic and endangered plant Phoebe chekiangensis. Biochem. SystEecol. 63, 183–189 (2015).
Ding, W. et al. Essential oil extracted from leaf of Phoebe bournei (Hemsl.) yang: chemical constituents, antitumor, antibacterial, hypoglycemic activities. Nat. Prod. Res. 32, 316–321 (2018).
Ye, Z. H. et al. Propagation, maintenance and landscape application of four ornamental plant species of Phoebe. Guangdong Landsc. Architecture 38, 48–51 (2016).
Chen, Z. et al. Effects of elevated ozone levels on photosynthesis, biomass and nonstructural carbohydrates of Phoebe bournei and Phoebe zhennan in subtropical China. Front. Plant Sci. 226, 310–317 (2018).
Li, T. & Min, X. Dormancy characteristics and germination requirements of Phoebe bournei seed. Sci. Hortic. 260, 108903 (2019).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness. Methods Mol. Biol. 1962, 227–245 (2019).
Massoni, J., Couvreur, T. L. P. & Sauquet, H. Five major shifts of diversification through the long evolutionary history of Magnoliidae (angiosperms). BMC Evol. Biol. 15, 49 (2015).
Soltis, D. E. & Soltis, P. S. Nuclear genomes of two magnoliids. Nat. Plants 5, 6–7 (2019).
Bai, G. et al. Genome-wide identification, gene structure and expression analysis of the MADS-Box gene family indicate their function in the development of tobacco (Nicotiana tabacum L.). Int. J. Mol. Sci. 20, 5043 (2019).
Colombo, M. et al. AGL23, a type I MADS‐box gene that controls female gametophyte and embryo development in Arabidopsis. Plant J. 54, 1037–1048 (2008).
Portereiko, M. F. et al. AGL80 is required for central cell and endosperm development in Arabidopsis. Plant Cell 18, 1862–1872 (2006).
Steffen, J. G. et al. AGL61 interacts with AGL80 and is required for central cell development in Arabidopsis. Plant Physiol. 148, 259–268 (2008).
Adamczyk, B. J. & Fernandez, D. E. MIKC* MADS domain heterodimers are required for pollen maturation and tube growth in Arabidopsis. Plant Physiol. 149, 1713–1723 (2009).
Liu, Y. et al. Functional conservation of MIKC*-Type MADS box genes in Arabidopsis and rice pollen maturation. Plant Cell 25, 1288–1303 (2013).
Hu, L. & Liu, S. Genome-wide analysis of the MADS-box gene family in cucumber. Genome 55, 245–256 (2012).
Lu, H. C., Liu, Z. J. & Lan, S. R. Genome sequencing reveals the role of MADS-box gene families in the floral morphology evolution of orchids. Hort. Plant J. 5, 240–246 (2019).
Arora, R. et al. MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics 8, 242 (2007).
Nesi, N. et al. The TRANSPARENT TESTA16 locus encodes the ARABIDOPSIS BSISTER MADS domain protein and is required for proper development and pigmentation of the seed coat. Plant Cell. 14, 2463–2479 (2002).
Jain, M. et al. The auxin-responsive GH3 gene family in rice (Oryza sativa). Funct. Gntegr. Genom. 6, 36–46 (2006).
Guuilfoyle, T. J. Auxin-regulated genes and promoters. in Biochemistry and Molecular Biology of Plant Hormones. (eds Hooykaas, P. J. J., Hall, M. A. & Libbenga, K. R.) 423–459 (Elsevier, Amsterdam, The Netherlands, 1999).
Vielba, J. M. Identification and initial characterization of a new subgroup in the GH3 gene family in woody plants. J. Plant Biochem. Biot. 28, 280–290 (2018).
Kong, W. et al. Evolutionary analysis of GH3 genes in six Oryza species/subspecies and their expression under salinity stress in Oryza sativa ssp. japonica. Plants 8, 30 (2019).
Park, J. E. et al. GH3-mediated auxin homeostasis links growth regulation with stress adaptation response in Arabidopsis. J. Biol. Chem. 282, 10036–10046 (2007).
Staswick, P. E. et al. Jasmonate response locus JAR1 and several related Arabidopsis genes encode enzymes of the firefly luciferase superfamily that show activity on jasmonic, salicylic, and indole-3-acetic acids in an assay for adenylation. Plant Cell. 14, 1405–1415 (2002).
Okrent, R. A., Brooks, M. D. & Wildermuth, M. C. Arabidopsis GH3.12 (PBS3) conjugates amino acids to 4-substituted benzoates and is inhibited by salicylate. J. Biol. Chem. 284, 9742–9754 (2009).
Staswick, P. E. Characterization of an Arabidopsis enzyme family that conjugates amino acids to indole-3-acetic acid. Plant Cell. 17, 616–627 (2005).
Sadaf, K. & Julie, M. S. Arabidopsis thaliana GH3.9 influences primary root. growth. Planta 226, 21–34 (2007).
Hsieh, H. L. et al. FIN219, an auxin-regulated gene, defines a link between phytochrome A and the downstream regulator COP1 in light control of Arabidopsis development. Gene Dev. 14, 1958–1970 (2000).
Mcclure, B. A. & Guilfoyle, T. J. Characterization of a class of small auxin-inducible soybean polyadenylated RNAs. Plant Mol. Biol. 9, 611–623 (1987).
Sato, A. et al. Light-dependent gravitropism and negative phototropism of inflorescence stems in a dominant Aux/IAA mutant of Arabidopsis thaliana, axr2. J. Plant Res. 127, 627–639 (2014).
Atamian, H. S. et al. Circadian regulation of sunflower heliotropism, floral orientation, and pollinator visits. Plant Sci. 353, 587–590 (2016).
Tohge, T., Watanabe, M., Hoefgen, R. & Fernie, A. R. Shi- kimate and phenylalanine biosynthesis in the green lineage. Front. Plant Sci. 4, 62 (2013).
Weng, J. K., Li, X., Bonawitz, N. D. & Chapple, C. Emerging strategies of lignin engineering and degradation for cellulosic biofuel production. Curr. Opin. Biotechnol. 19, 166–172 (2008).
Wilkins, O. et al. Expansion and diversification of the Populus R2R3-MYB family of transcription factors. Plant Physiol. 149, 981–993 (2009).
Kim, W. C. et al. Identification of direct targets of transcription factor MYB46 provides insights into the transcriptional regulation of secondary wall biosynthesis. Plant Mol. Biol. 85, 589–599 (2014).
McCarthy, R. L. et al. The poplar MYB transcription factors, PtrMYB3 and PtrMYB20, are involved in the regulation of secondary wall biosynthesis. Plant Cell Physiol. 51, 1084–1090 (2010).
Zhong, R., McCarthy, R. L., Haghighat, M. & Ye, Z. H. The poplar MYB master switches bind to the SMRE site and activate the secondary wall biosynthetic program during wood formation. PLoS ONE 8, e69219 (2013).
Xie, M. et al. A 5-Enolpyruvylshikimate 3-phosphate synthase functions as a transcriptional repressor in Populus. Plant Cell 30, 1645–1660 (2018).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Kent, W. J. BLAT—the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Jurka, J. et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenetic Genome Res. 110, 462–467 (2005).
Maja, T. G. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics 25, 4.10.1–4.10.14 (2009).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Zhao, X. & Hao, W. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Slater, G. S. C. et al. Automated generation of heuristics for biological sequence comparison. BMC Bioinforma. 6, 31 (2005).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Johnson, A. D. et al. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 24, 2938–2939 (2008).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinforma. 12, 491–491 (2011).
Altschul, S. F. et al. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL. Nucleic Acids Res. 31, 365–370 (2003).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Koonin, E. V. et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 5, R7 (2004).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Sam, G. J. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, 121–124 (2005).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009).
Li, L. et al. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Robert, C. E. et al. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Zhang, G. Q. et al. The Apostasia genome and the evolution of orchid. Nature 549, 379–383 (2017).
Maasorni, J. et al. Fossil calibration of Magnoliidae, an ancient lineage of angiosperms. Palaeontol Electron. 18, 1–25 (2015).
De, B. T., Cristianini, N., Demuth, J. P. & Hahn, M. W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006).
Tang, H., Krishnakumar, V. & Li, J. P. JCVI: JCVI Utility Libraries. https://github.com/tanghaibao/jcvi (2015).
Wang, K. et al. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 44, 1098–1103 (2012).
Blanc, G. & Wolfe, K. H. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell. 16, 1667–1678 (2004).
Alexandros, S. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 9, 9 (2014).
Hu, M. J. et al. Chromosome-scale assembly of the Kandelia obovata genome. Hortic. Res. 7, 75 (2020).
Kong, W. L. et al. Comparative genomic and transcriptomic analysis suggests the evolutionary dynamic of GH3 genes in Gramineae crops. Front. Plant Sci. 10, 1297 (2019).
Mukesh, J. et al. Genome-wide analysis, evolutionary expansion, and expression of early auxin-responsive SAUR gene family in rice (Oryza sativa). Genomics 88, 360–371 (2006).
Eddy, S. R. Accelerated profile HMM searches. PLoS Comput. Biol. 7, e1002195 (2011).
Letunic, I., Doerks, T. & Bork, P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 43, D257–D260 (2015).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Wang, Y. et al. GSA: genome sequence archive. Genom. Proteom. Bioinf. 15, 14–18 (2017).
Zhang, Z. et al. Database resources of the BIG data center in 2019. Nucleic Acids Res. 47, D8–D14 (2019).
Acknowledgements
This research was jointly funded by the Project of Forestry Peak Discipline at Fujian Agriculture and Forestry University, China (118/712018007); the Science and Technology Innovation Special Fund at Fujian Agriculture and Forestry University (118/KF2015088); and the 6th Project of Forest Seeding Breaking in Fujian Province ([2–19]357).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, SP., Sun, WH., Xiong, YF. et al. The Phoebe genome sheds light on the evolution of magnoliids. Hortic Res 7, 146 (2020). https://doi.org/10.1038/s41438-020-00368-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-020-00368-z
This article is cited by
-
Genome wide investigation of Hsf gene family in Phoebe bournei: identification, evolution, and expression after abiotic stresses
Journal of Forestry Research (2024)
-
Genome and whole-genome resequencing of Cinnamomum camphora elucidate its dominance in subtropical urban landscapes
BMC Biology (2023)
-
E. urophylla × E. grandis high-quality genome and comparative genomics provide insights on evolution and diversification of eucalyptus
BMC Genomics (2023)
-
Chloranthus genome provides insights into the early diversification of angiosperms
Nature Communications (2021)
-
The Chloranthus sessilifolius genome provides insight into early diversification of angiosperms
Nature Communications (2021)
