
Abstract
Horticultural plants play various and critical roles for humans by providing fruits, vegetables, materials for beverages, and herbal medicines and by acting as ornamentals. They have also shaped human art, culture, and environments and thereby have influenced the lifestyles of humans. With the advent of sequencing technologies, there has been a dramatic increase in the number of sequenced genomes of horticultural plant species in the past decade. The genomes of horticultural plants are highly diverse and complex, often with a high degree of heterozygosity and a high ploidy due to their long and complex history of evolution and domestication. Here we summarize the advances in the genome sequencing of horticultural plants, the reconstruction of pan-genomes, and the development of horticultural genome databases. We also discuss past, present, and future studies related to genome sequencing, data storage, data quality, data sharing, and data visualization to provide practical guidance for genomic studies of horticultural plants. Finally, we propose a horticultural plant genome project as well as the roadmap and technical details toward three goals of the project.
Similar content being viewed by others
Introduction
Horticultural plants mostly comprise vegetable-producing, fruit-bearing, ornamental, and beverage-producing plants and herbal medicinal plants. These plants have played important economic and social roles in the human lives and health by providing basic food needs, beautifying urban and rural landscapes, and improving personal esthetics. For example, the Food and Agriculture Organization of the United Nations reported that, while worldwide cereal food together is valued at 125 points (normalized value), vegetables and fruits together are valued at 137 points (http://faostat.fao.org). Horticultural plants also contribute to ecological balance by improving our biological environment by providing oxygen and balancing urban temperatures.
Horticultural plants are distributed among a wide variety of taxonomic plant spectra, which include a large number of flowering plants and a few early-divergent land plants. The sizes of their genomes vary greatly. For example, the vegetable garlic (Allium sativum) has a diploid genome (2n = 16) with an estimated genome size of >30 Gb1, and onion (Allium cepa) has a similar genome size2. In addition, most horticultural plants are domesticated, and their genome sequences have experienced strong artificial selection. For example, grape was found to have been cultivated (via viticulture) for >6000 years3; citrus, >4000 years4. In addition, some horticultural plants are intermediates of domesticated and wild plants, such as medicinal plants including ginseng (Panax ginseng), noto ginseng (Panax notoginseng), and Artemisia (Artemisia annua). Many domesticated horticultural plants have high levels of genetic diversity and heterozygosity, such as sunflower (10% of bases differ between homologous chromosomes)5, grape (7%)6, and potato (4.8%)7.
De novo sequencing of horticultural plant genomes
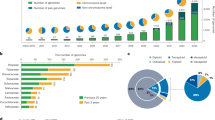
As of December 31, 2018, the genomes of 181 horticultural species have been sequenced (Table 1). These include 4 beverage, 47 fruit, 44 medicinal, 44 ornamental, and 42 vegetable plants (Fig. 1a). In terms of taxonomic distribution, these plants include 175 angiosperms, 2 gymnosperms, 3 lycophytes, and 1 moss (Fig. 1b). As shown in Fig. 1c, the number of sequenced genomes of horticultural plants completed each year has significantly increased from 1 in 2007 to 40 in 2018. Although most of the horticultural plants are angiosperms, the genome sequencing of non-angiosperm species has also demonstrated steady growth (Fig. 1c). Vegetables and fruits have been a focus of plant research in the past few years. However, only two vegetables and seven fruits had their genomes sequenced in 2018 (Fig. 1d). This is probably because many economically important vegetables and fruits were already sequenced prior to 2018.

a Distribution of genome-sequenced horticultural plants. b Botanical distribution of genome-sequenced horticultural plants. c Annual increase in the genome-sequenced horticultural plants by botanical taxonomy. d Annual increase in the genome-sequenced horticultural plants by horticultural category. e The reported 181 horticultural plant species fall into 30 angiosperm orders. f List of the released but not reported horticultural plant species
Some angiosperms have a significant role in the economy8. The 181 horticultural plants with sequenced genomes are distributed in 30 of the 64 angiosperm orders. Among these 30 orders, 7 (Fabales, Rosales, Cucurbitales, Brassicales, Sapindales, Solanales, and Laminales) have >10 species whose genomes have been sequenced (Fig. 1e), suggesting their vital importance to humans.
Most of the genome-sequenced plants fall into the Rosaceae family, which is a medium-sized family with approximately 4800 species (http://www.theplantlist.org), including many popular fruit-bearing and ornamental plants. The genome-decoded fruit-producing species include breadnut (Artocarpus camansi)9, ficus (Ficus carica)10, jujube (Ziziphus jujuba)11, strawberry and its close relatives (Fragaria × ananassa, Fragaria iinumae, Fragaria nipponica, Fragaria nubicola, Fragaria orientalis, Fragaria vesca)12,13,14, apple (Malus domestica)15, morus (Morus notabilis)16, sweet cherry (Prunus avium)17, peach (Prunus persica)18, Chinese pear (Pyrus bretschneideri)19, European pear (Pyrus communis)20, and black raspberry (Rubus occidentalis)21. The genome-decoded ornamentals include mei (Prunus mume)22, sakura (Prunus yedoensis)23, and rose and its close relatives (Rosa × damascene, Rosa chinensis, Rosa multiflora, and Rosa roxburghii)24,25,26. However, the genomes of many important fruit-bearing Rosales plants, such as Crataegus pinnatifida, Malus prunifolia, Eriobotrya japonica, Armeniaca vulgaris, and Prunus salicina, and of Rosales ornamentals, such as Photinia serrulata, Spiraea thunbergii, Cotoneaster multiflorus, and Rubus japonicas, have not yet been sequenced. The available genome sequences of Rosales species have largely improved our understanding of the biology of fruits and flowers. For example, the high-quality apple genome sequence showed that a single allele is responsible for red fruit peal coloration27, and the reference genome of rose has provided insights into the floral color and scent pathways25.
The Solanaceae family consists of ~2700 species (http://www.theplantlist.org) that include a number of vegetable, medicinal, and ornamental species. The genomes of several important Solanaceae vegetable species have been sequenced, such as tomato (Solanum lycopersicum, Solanum pimpinellifolium)28,29, potato (Solanum tuberosum)30, pepper (Capsicum annuum, Capsicum baccatum, Capsicum chinense)31,32,33, and eggplant (Solanum melongena)34. Solanaceae ornamentals include ivy morning glory (Ipomoea nil)35, ornamental tobacco (Nicotiana sylvestris)36, and petunia (Petunia axillaris, Petunia inflate)37. Although these genomes have helped to understand the evolution of Solanaceae plants, additional Solanaceae horticultural genomes need to be sequenced. These include the sequences of the medicinal plants Datura arborea, Datura metel, and Datura innoxia and the ornamentals Petunia spp., Nicotiana spp., Lycium spp., Solanum spp., Cestrum spp., Calibrachoa spp., and Solandra spp. These available genome sequences have helped to decipher the evolution and genomic basis of metabolites such as vitamin C (or ascorbic acid)38 in tomato and alkaloids in tobacoo39.
The Fabaceae family, consisting of ~19,000 known species, is the third largest angiosperm family by number of species richness, followed by the Orchidaceae and Asteraceae families. Although only dozens of Fabaceae genomes have been sequenced8, many of them are from horticultural species. The genome-decoded Fabaceae vegetable plants include pigeon pea (Cajanus cajan)40, chickpea and its relative (Cicer arietinum, Cicer reticulatum)41,42, soybean (Glycine max)43, barrelclover (Medicago truncatula)44, common bean (Phaseolus vulgaris)45, faba bean (Vicia faba)46, adzuki bean (Vigna angularis)47, and mung bean (Vigna radiata)48. The genome-sequenced Fabaceae ornamentals include eastern redbud (Cercis canadensis)49, narrowleaf lupin (Lupinus angustifolius)50, and mimosa (Mimosa pudica). The Fabaceae medicinal plants with sequenced genomes include Chinese uralensis (Glycyrrhiza uralensis)51 and red clover (Trifolium pratense)52. Legumes are considered a valuable source of food in the future53; thus the sequencing of their genomes would be valuable. Determining the genomic basis of legume–rhizobium interactions would help not only to solve a classic fundamental problem in biology but also to improve nitrogen utilization in horticultural plants.
The Brassicaceae family is a medium-sized family with ~4000 species, including many horticultural plant species. The Brassicaceae vegetable plants with sequenced genomes include Zhacai (Brassica juncea)54, cabbage (Brassica oleracea)55, napa cabbage (Brassica rapa)56, Capsella (Capsella bursa-pastoris and Capsella rubella)57,58, radish (Raphanus sativus)59, and field pennycress (Thlaspi arvense)60. The genomes of the Brassicaceae medicinal plants Eutrema yunnanense61 and maca (Lepidium meyenii)62 have also been sequenced. With these genome sequences at hand, the genomic features of common ancestors and the subsequent evolution of the Brassicaceae can be clarified, such as the intron evolution within the Brassicaceae63, and gene and genome duplication events within the Brassicaceae64,65. These genomes would also shed light on the evolution of the hypocotyl, as has been reported in maca62 and radish59. Within the Brassicaceae family, we could foresee a growing demand for the genome sequencing of horticultural Brassicaceae plants, both for evolutionary research and for decoding the molecular basis of economically important traits.
The Cucurbitaceae family includes >3700 species belonging to 134 genera (www.theplantlist.org). Within this family, the genome-decoded vegetable plants include silver-seed gourd (Cucurbita argyrosperma)66, winter squash (Cucurbita maxima)67, pumpkin (Cucurbita moschata)67, summer squash (Cucurbita pepo)68, bottle gourd (Lagenaria siceraria)69, and bitter melon (Momordica charantia)70. The genome-decoded fruit species include muskmelon (Cucumis melo)71 and watermelon (Citrullus lanatus)72. The only genome-decoded medicinal plant is monk fruit (Siraitia grosvenorii)73,74. Via analysis of these available genome sequences, it was found that a tetraploid-inducing event occurred in the last common ancestor of the Cucurbitaceae species75. These genome sequences can also help to better understand the domestication history76 and fruit development77. Increasing numbers of the wild relatives of these economically important crop species, as well as those of thousands of plant cultivars, will be sequenced in the near future, providing additional details and surprises.
The Rutaceae or citrus family consists of 158 genera and 6686 species (www.theplatlist.org). The Rutaceae fruit-bearing plants with sequenced genomes include clementine (Citrus clementina)78, pomelo (Citrus grandis)79, Ichang papeda (Citrus ichangensis)79, citrumelo (Citrus paradisi × Poncirus trifoliate)80, mandarin orange (Citrus reticulata)81, sweet orange (Citrus sinensis)82, and cold-hardy mandarin (Citrus unshiu)83. The Rutaceae medicinal plants with sequenced genomes include jiu bing le (Atalantia buxifolia)79 and citron (Citrus medica)79. Via analysis of these genome sequences, the evolutionary origin and evolutionary changes in the Citrus genus during domestication were mapped84. In the future, the genome sequences of Rutaceae fruit-bearing plants including lemon (Citrus limon), calamansi (Citrofortunella microcarpa), lime (Citrus spp. hybrids), kumquat (Citrus japonica), and grapefruit (Citrus × paradisi) will require genome sequencing.
Genome resequencing and the pan-genome of horticultural plants
A single reference genome sequence is not sufficient for identifying the best candidate genes for molecular breeding or for understanding the genomic background of a population due to the prevalence of genomic structural variations. Compared to the construction of a reference genome, genome resequencing usually requires less sequencing coverage. It is feasible to obtain a high-quality resequenced genome via mapping to a reference genome. A pan-genome is the summary of genomes of a species obtained by comparing a large number of resequenced genomes of a species or, occasionally, a genus. A pan-genome can help to understand the size of a core genome (defined as the conserved part among the related genomes), the size of a pan-genome, and the amount and nature of variations within a species or a genus, which improve our understanding of the evolution of a species/genu, as well as of agronomic traits. Currently, a growing number of pan-genomes among horticultural plants have been constructed (Table 2).
Soybean is an economically important vegetable crop; in addition to being a source of human protein, it is an important source of vegetable oil. Glycine soja is the closest wild relative to cultivated soybean (Glycine max). The G. soja pan-genome was the first horticultural pan-genome released, which occurred in 2014 and consisted of seven wild accessions85 (Table 2). This pan-genome revealed that, when more genomes were added, the number of shared genes decreased, and in contrast, the number of total genes increased when more genomes were added. In addition, this pan-genome confirmed that a single reference genome does not adequately represent the genomic and genetic diversity of a species. Because the reference genome of G. soja was not previously available, those researchers assembled all seven genomes with the de novo assembly method, but this method was not adopted by subsequent researchers.
Assembly of the B. oleracea pan-genome86 is another early trial in the genomic research of horticultural plants (Table 2). It is relatively small, created using nine morphologically diverse varieties (covering two cabbage, one broccoli, one brussels sprout, one kohlrabi, two cauliflowers, and one kale plant) and a wild relative, Brassica macrocarpa. Through the analyses of this pan-genome, we observed that 20% of genes are absent in some cultivar(s), and there are presence–absence variations (PAVs), including those related to major agronomic traits. This is a pioneering study that provided assembled pan-genome contigs, pan-genome annotations, and the GBrowse tool, available at http://brassicagenome.net.
Pepper plants are important vegetable plants with distinct fruit morphologies. The pepper pan-genome has been generated for the pepper genus Capsicum87. This pan-genome consists of 5 species and 383 cultivars, all of which have 15 chromosomes. In addition to the comparison of PAVs among this large amount of pepper cultivars, the pan-genome is also useful in linking the association between important agronomic traits and corresponding genes. These valuable pan-genome data and JBrowse and other search tools are available (www.pepperpan.org:8012).
Sunflower plants provide seed that can be used for cooking oil and serve as popular ornamentals. The sunflower pan-genome was created by sequencing 493 accessions, including cultivars, landraces, and wild relatives5. A total of 61,205 genes have been identified within the gene set of the sunflower pan-genome. Via the aid of this pan-genome, the understanding of the evolutionary history of sunflower species has significantly improved, and genes linked to biotic stress resistance have been identified5. Although pan-genome data can be found in the sunflower genome database (www.sunflowergenome.org), no publicly accessible tool has been built to date (accessed March 31, 2019).
Reference genome sequences are necessary to identify genes and to understand evolutionary trajectory. However, a pan-genome can help to uncover additional details. For example, relying on the tomato genome sequence, researchers mapped only several genes and pathways controlling fruit ripening28. These flesh- and flavor-related genes are the best targets in breeding. Moreover, genome sequences allow comprehensive and systematic analyses of fruit biology. Furthermore, via the sequencing of a tomato population and analysis of its pan-genome consisting of 725 accessions, the genes selected during domestication and quality improvement were identified88. Thus a pan-genome not only improves our understanding of crop evolution but also is useful for the discovery of novel genes and breeding.
Data storage and visualization
In addition to comprehensive plant-centric databases such as Phytozome (https://phytozome.jgi.doe.gov) and EnsemblPlants (http://plants.ensembl.org), 27 horticultural plant-specific genome databases have been constructed (Table 3). Among these, 22 provide data for downloading. Some databases are freely accessible to all users, while others provide only limited access to specific data or users. For example, the Genome Database for Rosaceae89 requires user registration and a login to access the breeding data.
Visualization of genomic data of horticultural plants is challenging due to the heterogeneous nature of the different types of data. GBrowse90 and JBrowse91,92,93 are powerful tools that provide a visualization of various levels of genomic features. The availability of genomic analysis tools also varies greatly among databases. BLAST-related tools such as NCBI-BLAST94 and viroBLAST95 are provided by some databases for homologous sequence searches and sequence comparisons. Gene query tools can help to obtain details of genes such as their sequence, annotation, and expression. HMMER96 searches allow the inference and extraction of gene families from genomes in the database. Syntenic tools allow the identification and visualization of genome-wide syntenic relationships across genomes. The BioCyc tools (https://biocyc.org) allow users to navigate individual pathways or the whole metabolic map of a genome for functional analyses97.
The Genome Database for Rosaceae (GDR), which was developed by the main bioinformatics laboratory at Washington State University89, is well known among the Rosaceae research community and even the plant research community. It covers the genome sequences of 18 Rosaceae species (Fragaria vesca, F. ananassa, F. iinumae, F. nipponica, F. nubicola, F. orientalis, Malus domestica, Potentilla micrantha, Prunus avium, Prunus domestica, Prunus dulcis, Prunus persica, Prunus yedoensis, Pyrus bretschneideri, Pyrus communis, Rosa chinensis, Rosa multiflora, and Rubus occidentalis), which are categorized into seven genera: Fragaria, Malus, Potentilla, Prunus, Pyrus, Rosa, and Rubus. To facilitate online analyses, a series of tools are provided, including genomic tools (BLAST+, JBrowse, Primer3, Sequence Retrieval, MapViewer, Synteny Viewer), metabolomic tools (GDRcyc, Pathway Inspector), and breeding tools (Breeding information Management System (BMS), Breeders Toolbox). The same team at Washington State University also developed a series of horticultural plant-themed databases, including the Citrus Genome Database, Cool-Season Food Legume Crop Database resources, and Genome Database for Vaccinium (GRIN). All these databases share a similar data process standard and have built-in bioinformatics tools.
The Sol Genomics Network (SGN)98, a database of Solanaceae genomic and phenotypic data and tools, was developed by Mueller’s team from the Boyce Thompson Institute for Plant Research and Cornell University. The SGN includes 11 genomes: those of Solanum lycopersicum, S. lycopersicoides, S. pimpinellifolium, S. tuberosum, S. pennellii, Capsicum annuum, Nicotiana attenuata, N. benthamiana, N. tabacum, Petunia axillaris, and P. inflata. These species are categorized into four economically important genera: Solanum, Capsicum, Nicotiana, and Petunia. For online analyses of genomic sequences, BLAST, Alignment Analyzer, Tree Browser, and VIGS tools are available. For mapping of various data, JBrowse, Comparative Map Viewer, CAPS Designer, and solQTL are provided. Some tools have been developed for common molecular wet laboratory experiments, including In-Silico PCR, the Tomato Expression Atlas, and the Tomato Expression Database. Systems biology tools such as SolCyc Biochemical Pathways99, Coffee Interactome Data, and the SGN Ontology Browser are provided. The Breeders Toolbox was developed for breeders. The same team also developed a series of horticultural plant-themed databases, including the YamBase (https://yambase.org), CassavaBase (https://cassavabase.org), and MusaBase (https://musabase.org) databases. All these databases adhere to the release of genomic data before publication (the Toronto Agreement)100.
The Cucurbit Genomics Database (CuGenDB)101 currently hosts eight high-quality genome sequences corresponding to those of cucumber (Cucumis sativus), water melon (Citrullus lanatus), winter squash (Cucurbita maxima), pumpkin (Cucurbita moschata), summer squash (Cucurbita pepo), muskmelon (Cucumis melo), bottle gourd (Lagenaria siceraria), and silver-seed gourd (Cucurbita argyrosperma). The search and batch query system allow searching for sequences and annotations. To display genomic details, the JBrowse, BLAST, Gene Ontology (GO), Synteny Viewer, CAMP, and expression viewer tools are available. To display metabolic pathways, CucurbitCyc and Pathway enrichment tools are available.
The Brassica Database (BARD)102, a database of important Brassica species, covers the vegetable species Brassica rapa and B. oleracea, as well as the model plant Arabidopsis and Brassicaceae close relatives. In addition to its genomic data, the BRAD database hosts a curated list of genes involved with anthocyanins, resistance, auxin, flowering, and glucosinolates and a full list of gene families that are of considerable importance in Brassica research. BLAST and JBrowse tools were built for visualization of genomic data, and syntenic tools are useful for comparative analyses.
The Herbal Medicine Omics Database103 includes genomic, transcriptomic, pathway, and metabolomic data for medicinal plants, although the medicinal properties of some plants are recognized only in some parts of the world. In this database, hundreds of medicinal plants are included. However, the database currently provides only the BLAST and GBrowse tools for the visualization of omics data. Other collected omic data can be downloaded but cannot be analyzed or visualized online.
There are other tool-specific databases that can be very useful for the visualization and online analyses of horticultural plant genome sequences. The Plant Genome Duplication Database (PGDD)104 offers online analyses of gene synteny and visualization of different results, such as dot plots (macrosynteny) and local genomic comparison plots (microsynteny). The built-in Map-View tool allows mapping of a given sequence to the genomes of 47 species from the PGDD (data accessed on March 31, 2019). The Plant Duplicate Gene Database105 is a collection of 141 plant species and offers online analysis and visualization of duplicated genes in select species.
Discussions and future perspectives
The horticultural plant genome project
It is challenging to determine the exact number of species or cultivars that exist for horticultural plants. In terms of fruit-bearing plants, at least 91 species are economically important and produce fruit that are consumed (https://simple.wikipedia.org/wiki/List_of_fruits). More than 200 vegetable plants are consumed (https://simple.wikipedia.org/wiki/List_of_vegetables). The exact number of ornamentals is also unclear, as novel cultivars are produced each year. However, it has been estimated that there are >6000 ornamental cultivars (https://www.rhs.org.uk/plants/pdfs/agm-lists/agm-ornamentals-(1).pdf), and many cultivars are created and disappear each year. Up to December 2018, genome sequences had been decoded for only 181 species, accounting for only a small proportion of the total horticultural plant species. Hence, there is a strong need to sequence additional genomes for more horticultural plants that would be valuable for comparative genomics, to better understand their evolutionary history, and to possibly make genetic modifications to better utilize these plant species.
Here we propose a horticultural plant genome project (HPGP) with three goals (Fig. 2). The first goal of the HPGP is to generate reference genome sequences for all horticultural plants, after which pan-genomes and core collections would be generated as genetic banks for horticultural plants. Two recently developed genome assembly methods could be applied to decode highly ploidy71 and highly heterozygous106,107,108 horticultural genomes. The second goal is to identify the various genomic variations within a pan-genome. In addition, the mechanistic signatures leading to the variations would be explored. The third goal is to link the phenotypes to the genomic regions. Two methods would be applied: quantitative trait locus methods to correlate genomic variations with a quantitative trait and genome-wide association study methods to associate genomic variation with many genomic variations from different individuals109,110. The good news is that the Earth Genome Project and the 1000-Plant Genome Project will accelerate the genome sequencing process of horticultural plants.

The first goal of HPGP is to generate all reference genome sequences for horticultural plants, after which pan-genomes and core collections will be generated as a gene bank for horticultural plants. Two recently developed methods would be applied to decode the highly ploidy and highly heterozygous horticultural genomes. The second goal is to detect the various genomic variations within a pan-genome. In addition, the mechanistic signatures leading to the variations would be explored. The third goal is to link the phenotypes with the genomic regions. Two methods would be applied: the quantitative trait locus (QTL) method to correlate genomic variations with a quantitative trait and the genome-wide association study (GWAS) method to associate genomic variation with many genomic variations from different individuals ***p < 0.001
The timeline for obtaining the genome sequences of all horticultural plants at both draft and reference scales (goal one of the HPGP) would be short—within 3–5 years—because the cost for sequencing is dropping rapidly. However, collecting and sequencing the population definitely requires worldwide collaborations and would take >10 years. The second goal is to analyze the genomic variations to identify the mechanistic signatures within a population, which is also time consuming and would be gradually achieved. The third goal is an advanced step that occurs after or concurrently with the second goal. Although these last two goals appear to be enormous challenges, we are confident in the ability to achieve most of these two goals in model horticultural plants such as the tomato, cucumber, and strawberry in the coming years.
In addition, the quality of assembly and annotation of existing reference genomes of horticultural plants need to be further improved. Although a few tools such as BUSCO111 and CEGMA112 have been widely used to evaluate the quality of genome annotations, a good standard is still not available for the systematic evaluation of the quality of genome assemblies. As a result, the quality of the genome assemblies is very uneven and is sometimes related to the complexity or heterozygosity of the taxa. This situation is changing as sequencing platforms are being upgraded. For example, since the first apple genome sequence was released in 2010 based on next-generation sequencing technology15, an improved version produced by next-generation sequencing (NGS) and PacBio technologies was released in 2016113. The third improved version of the apple genome, which was obtained using a combination of NGS, PacBio, and Bionano technologies, was released in 2017114. The fourth improved version was released in 2019, based on the utilization of NGS, PacBio, and Hi-C technologies27. In the future, the quality of the reference genome should reach certain minimal standards upon which the community can agree, similar to the proposal for bacteria and archaea115, thereby leading to more accurate pan-genome analyses and biotechnology.
Storage and access of genomic data constitute another problem concerning horticultural biologists and bioinformatics scientists. For access to genome sequences and raw sequencing data, a number of public databases are usually the first choice of researchers due to the nature of their stability, low cost, and ease of access. The well-known public databases include the NCBI (https://ncbi.nlm.nih.gov), EMBL (www.embl.org), CNGB (www.cngb.org), BIGD (bigd.big.ac.cn), DDBJ (www.ddbj.nig.ac.jp), GigaDB (gigadb.org), Dryad (www.datadryad.org), and Phytozome (https://phytozome.jgi.doe.gov) databases. To share these data with worldwide researchers, we encourage the release of data before publication, as was suggested by the Toronto Agreement in 2009100.
The need for a horticultural plant-centric database
Unlike agricultural plants, horticultural plants share multiple features. For example, plant growth requires controlled conditions with specific equipment or facilities; plants generally need grafting, postharvest treatment, and a long juvenile phase; and plants usually undergo asexual reproduction and have unique specialized metabolism. All of these concerns make it hard to study these traits in model plants or via regular tools. Uniting the various omic data and the development of novel tools for horticultural plants are needed. Moreover, aside from the comprehensive plant databases and the 27 horticultural plant-specific databases mentioned above, there is still an increasing need to find and compare an increased amount of data for horticultural plants. However, horticultural biologists usually need to frequently deal with breeders; thus the need to create a comprehensive horticultural database to meet the interests of basic biologists and breeders is largely required. Such a database should cover as many horticultural plant genomes as possible and should provide an integrated set of bioinformatics tools. We believe that, in the future, the need for such a comprehensive database of all horticultural plants will satisfy additional horticulture researchers and breeders.
Given the advancement of sequencing technologies and reduced costs, the genome sequencing data of horticultural plants are accumulating rapidly. The storage, analyses, and sharing of large collections of genome sequencing data are becoming even more laborious and time consuming. The integrative analysis of various omic data, such as genomic, transcriptomic, metabolomic, phenomic, and breeding data, have become a major challenge for many horticultural biologists and requires coordinated efforts of scientists from different fields. For data processing and visualization, we recommend using BioMart tools, which could be easily built into a database. For database construction, we suggest following the template of the Tripal series (www.tripal.infor)8. Finally, we believe that, with a fostered collaboration of the horticultural community, the HPGP and subsequent knowledge and experiences will greatly benefit biology researchers and breeders.
References
Egea, L. A., Merida-Garcia, R., Kilian, A., Hernandez, P. & Dorado, G. Assessment of genetic diversity and structure of large garlic (Allium sativum) germplasm bank, by diversity arrays technology “genotyping-by-sequencing” platform (DArTseq). Front. Genet. 8, 98 (2017).
Peska, V., Mandakova, T., Ihradska, V. & Fajkus, J. Comparative dissection of three giant genomes: Allium cepa, Allium sativum, and Allium ursinum. Int. J. Mol. Sci. 20, E733 (2019).
Li, H. et al. The wolds of wine: old, new and ancient. Wine Econ. Pol. 7, 178–182 (2018).
Zheng, Z., Chen, J. & Deng, X. Historical perspectives, management, and current research of citrus HLB in Guangdong Province of China, where the disease has been endemic for over a hundred years. Phytopathology 108, 1224–1236 (2018).
Hubner, S. et al. Sunflower pan-genome analysis shows that hybridization altered gene content and disease resistance. Nat. Plants 5, 54–62 (2019).
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007).
Leisner, C. P. et al. Genome sequence of M6, a diploid inbred clone of the high-glycoalkaloid-producing tuber-bearing potato species Solanum chacoense, reveals residual heterozygosity. Plant J. 94, 562–570 (2018).
Chen, F. et al. The sequenced angiosperm genomes and genome databases. Front. Plant Sci. 9, 418 (2018).
Gardner, E. M., Johnson, M. G., Ragone, D., Wickett, N. J. & Zerega, N. J. Low-coverage, whole-genome sequencing of Artocarpus camansi (Moraceae) for phylogenetic marker development and gene discovery. Appl Plant Sci. 4, apps.1600017 (2016).
Mori, K. et al. Identification of RAN1 orthologue associated with sex determination through whole genome sequencing analysis in fig (Ficus carica L.). Sci. Rep. 7, 41124 (2017).
Liu, M. J. et al. The complex jujube genome provides insights into fruit tree biology. Nat. Commun. 5, 5315 (2014).
Edger, P. P. et al. Origin and evolution of the octoploid strawberry genome. Nat. Genet. 51, 541–547 (2019).
Shulaev, V. et al. The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 43, 109–116 (2011).
Hirakawa, H. et al. Dissection of the octoploid strawberry genome by deep sequencing of the genomes of Fragaria species. DNA Res. 21, 169–181 (2014).
Velasco, R. et al. The genome of the domesticated apple (Malus x domestica Borkh.). Nat. Genet. 42, 833–839 (2010).
He, N. et al. Draft genome sequence of the mulberry tree Morus notabilis. Nat. Commun. 4, 2445 (2013).
Shirasawa, K. et al. The genome sequence of sweet cherry (Prunus avium) for use in genomics-assisted breeding. DNA Res. 24, 499–508 (2017).
Verde, I. et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 45, 487–494 (2013).
Wu, J. et al. The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res. 23, 396–408 (2013).
Chagne, D. et al. The draft genome sequence of European pear (Pyrus communis L. ‘Bartlett’). PLoS ONE 9, e92644 (2014).
VanBuren, R. et al. A near complete, chromosome-scale assembly of the black raspberry (Rubus occidentalis) genome. Gigascience 7, giy094 (2018).
Zhang, Q. X. et al. The genome of Prunus mume. Nat. Commun. 3, 1318 (2012).
Baek, S. et al. Draft genome sequence of wild Prunus yedoensis reveals massive inter-specific hybridization between sympatric flowering cherries. Genome Biol. 19, 127 (2018).
Nakamura, N. et al. Genome structure of Rosa multiflora, a wild ancestor of cultivated roses. DNA Res. 25, 113–121 (2018).
Raymond, O. et al. The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 50, 772–777 (2018).
Lu, M., An, H. M. & Li, L. L. Genome survey sequencing for the characterization of the genetic background of Rosa roxburghii tratt and leaf ascorbate metabolism genes. PLoS ONE 11, e0147530 (2016).
Zhang, L. et al. A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat. Commun. 10, 1494 (2019).
Sato, S. et al. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012).
Razali, R. et al. The genome sequence of the wild tomato Solanum pimpinellifolium provides insights into salinity tolerance. Front. Plant Sci. 9, 1402 (2018).
Xu, X. et al. Genome sequence and analysis of the tuber crop potato. Nature 475, 189–194 (2011).
Qin, C. et al. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc. Natl Acad. Sci. USA 111, 5135–5140 (2014).
Kim, S. et al. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat. Genet. 46, 270–278 (2014).
Ahn, Y. K. et al. Whole genome resequencing of Capsicum baccatum and Capsicum annuum to discover single nucleotide polymorphism related to Powdery Mildew resistance. Sci. Rep. 8, 5188 (2018).
Hirakawa, H. et al. Draft genome sequence of eggplant (Solanum melongena L.): the representative solanum species indigenous to the old world. DNA Res. 21, 649–660 (2014).
Hoshino, A. et al. Genome sequence and analysis of the Japanese morning glory Ipomoea nil. Nat. Commun. 7, 13295 (2016).
Sierro, N. et al. Reference genomes and transcriptomes of Nicotiana sylvestris and Nicotiana tomentosiformis. Genome Biol. 14, R60 (2013).
Bombarely, A. et al. Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida. Nat. Plants 2, 16074 (2016).
Ruggieri, V., Bostan, H., Barone, A., Frusciante, L. & Chiusano, M. L. Integrated bioinformatics to decipher the ascorbic acid metabolic network in tomato. Plant Mol. Biol. 91, 397–412 (2016).
Sierro, N. et al. The tobacco genome sequence and its comparison with those of tomato and potato. Nat. Commun. 5, 3833 (2014).
Varshney, R. K. et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 30, 83–89 (2012).
Varshney, R. K. et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 31, 240–246 (2013).
Gupta, S. et al. Draft genome sequence of Cicer reticulatum L., the wild progenitor of chickpea provides a resource for agronomic trait improvement. DNA Res. 24, 1–10 (2017).
Schmutz, J. et al. Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183 (2010).
Young, N. D. et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480, 520–524 (2011).
Schmutz, J. et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 46, 707–713 (2014).
Cooper, J. W. et al. Enhancing faba bean (Vicia faba L.) genome resources. J. Exp. Bot. 68, 1941–1953 (2017).
Kang, Y. J. et al. Draft genome sequence of adzuki bean, Vigna angularis. Sci. Rep. 5, 8069 (2015).
Kang, Y. J. et al. Genome sequence of mungbean and insights into evolution within Vigna species. Nat. Commun. 5, 5443 (2014).
Griesmann, M. et al. Phylogenomics reveals multiple losses of nitrogen-fixing root nodule symbiosis. Science 361, eaat1743 (2018).
Hane, J. K. et al. A comprehensive draft genome sequence for lupin (Lupinus angustifolius), an emerging health food: insights into plant-microbe interactions and legume evolution. Plant Biotechnol. J. 15, 318–330 (2017).
Mochida, K. et al. Draft genome assembly and annotation of Glycyrrhiza uralensis, a medicinal legume. Plant J. 89, 181–194 (2017).
De Vega, J. J. et al. Red clover (Trifolium pratense L.) draft genome provides a platform for trait improvement. Sci. Rep. 5, 17394 (2015).
Cullis, C. & Kunert, K. J. Unlocking the potential of orphan legumes. J. Exp. Bot. 68, 1895–1903 (2017).
Yang, J. et al. The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 48, 1225–1232 (2016).
Liu, S. Y. et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5, 3930 (2014).
Wang, X. W. et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039 (2011).
Kasianov, A. S. et al. High-quality genome assembly of Capsella bursa-pastoris reveals asymmetry of regulatory elements at early stages ofpolyploid genome evolution. Plant J. 91, 278–291 (2017).
Slotte, T. et al. The Capsella rubella genome and the genomic consequences of rapid mating system evolution. Nat. Genet. 45, 831–835 (2013).
Kitashiba, H. et al. Draft sequences of the radish (Raphanus sativus L.) genome. DNA Res. 21, 481–490 (2014).
Dorn, K. M., Fankhauser, J. D., Wyse, D. L. & Marks, M. D. A draft genome of field pennycress (Thlaspi arvense) provides tools for the domestication of a new winter biofuel crop. DNA Res. 22, 121–131 (2015).
Guo, X. et al. The genomes of two Eutrema species provide insight into plant adaptation to high altitudes. DNA Res. https://doi.org/10.1093/dnares/dsy003 (2018).
Zhang, J. et al. Genome of plant maca (Lepidium meyenii) illuminates genomic basis for high-altitude adaptation in the central Andes. Mol. Plant 9, 1066–1077 (2016).
Milia, G., Camiolo, S., Avesani, L. & Porceddu, A. The dynamic loss and gain of introns during the evolution of the Brassicaceae. Plant J. 82, 915–924 (2015).
Singh, S., Das, S. & Geeta, R. A segmental duplication in the common ancestor of Brassicaceae is responsible for the origin of the paralogs KCS6-KCS5, which are not shared with other angiosperms. Mol. Phylogenet. Evol. 126, 331–345 (2018).
Murat, F. et al. Understanding Brassicaceae evolution through ancestral genome reconstruction. Genome Biol. 16, 262 (2015).
Barrera-Redondo, J. et al. The genome of Cucurbita argyrosperma (silver-seed gourd) reveals faster rates of protein-coding gene and long noncoding RNA turnover and neofunctionalization within Cucurbita. Mol. Plant 12, 506–520 (2019).
Sun, H. et al. Karyotype stability and unbiased fractionation in the paleo-allotetraploid cucurbita genomes. Mol. Plant 10, 1293–1306 (2017).
Montero-Pau, J. et al. De novo assembly of the zucchini genome reveals a whole-genome duplication associated with the origin of the Cucurbita genus. Plant Biotechnol. J. 16, 1161–1171 (2018).
Wu, S. et al. The bottle gourd genome provides insights into Cucurbitaceae evolution and facilitates mapping of a Papaya ring-spot virus resistance locus. Plant J. 92, 963–975 (2017).
Urasaki, N. et al. Draft genome sequence of bitter gourd (Momordica charantia), a vegetable and medicinal plant in tropical and subtropical regions. DNA Res. 24, 51–58 (2017).
Garcia-Mas, J. et al. The genome of melon (Cucumis melo L.). Proc. Natl Acad. Sci. USA 109, 11872–11877 (2012).
Guo, S. et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 45, 51–58 (2013).
Itkin, M. et al. The biosynthetic pathway of the nonsugar, high-intensity sweetener mogroside V from Siraitia grosvenorii. Proc. Natl Acad. Sci. USA 113, E7619–E7628 (2016).
Xia, M. et al. Improved de novo genome assembly and analysis of the Chinese cucurbit Siraitia grosvenorii, also known as monk fruit or luo-han-guo. Gigascience 7, giy067 (2018).
Wang, J. et al. An Overlooked paleotetraploidization in Cucurbitaceae. Mol. Biol. Evol. 35, 16–26 (2018).
Yang, L. M. et al. Chromosome rearrangements during domestication of cucumber as revealed by high-density genetic mapping and draft genome assembly. Plant J. 71, 895–906 (2012).
Shang, Y. et al. Biosynthesis, regulation, and domestication of bitterness in cucumber. Science 346, 1084–1088 (2014).
Wu, G. A. et al. Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nat. Biotechnol. 32, 656–662 (2014).
Wang, X. et al. Genomic analyses of primitive, wild and cultivated citrus provide insights into asexual reproduction. Nat. Genet. 49, 765–772 (2017).
Zhang, Y., Barthe, G., Grosser, J. W. & Wang, N. Transcriptome analysis of root response to citrus blight based on the newly assembled Swingle citrumelo draft genome. BMC Genomics 17, 485 (2016).
Wang, L. et al. Genome of wild mandarin and domestication history of mandarin. Mol. Plant 11, 1024–1037 (2018).
Xu, Q. et al. The draft genome of sweet orange (Citrus sinensis). Nat. Genet. 45, 59–66 (2013).
Shimizu, T. et al. Draft sequencing of the heterozygous diploid genome of satsuma (Citrus unshiu Marc.) using a hybrid assembly approach. Front. Genet. 8, 180 (2017).
Wu, G. A. et al. Genomics of the origin and evolution of Citrus. Nature 554, 311–316 (2018).
Li, Y. H. et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 32, 1045–1052 (2014).
Golicz, A. A. et al. The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun. 7, 13390 (2016).
Ou, L. J. et al. Pan-genome of cultivated pepper (Capsicum) and its use in gene presence-absence variation analyses. New Phytol. 220, 360–363 (2018).
Gao, L. et al. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 51, 1044–1051 (2019).
Jung, S. et al. The Genome Database for Rosaceae (GDR): year 10 update. Nucleic Acids Res. 42, D1237–D1244 (2014).
Stein, L. D. Using GBrowse 2.0 to visualize and share next-generation sequence data. Brief Bioinform. 14, 162–171 (2013).
Westesson, O., Skinner, M. & Holmes, I. Visualizing next-generation sequencing data with JBrowse. Brief Bioinform. 14, 172–177 (2013).
Hofmeister, B. T. & Schmitz, R. J. Enhanced JBrowse plugins for epigenomics data visualization. BMC Bioinformatics 19, 159 (2018).
Buels, R. et al. JBrowse: a dynamic web platform for genome visualization and analysis. Genome Biol. 17, 66 (2016).
Johnson, M. et al. NCBI BLAST: a better web interface. Nucleic Acids Res. 36, W5–W9 (2008).
Deng, W., Nickle, D. C., Learn, G. H., Maust, B. & Mullins, J. I. ViroBLAST: a stand-alone BLAST web server for flexible queries of multiple databases and user’s datasets. Bioinformatics 23, 2334–2336 (2007).
Potter, S. C. et al. HMMER web server: 2018 update. Nucleic Acids Res. 46, W200–W204 (2018).
Caspi, R. et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 46, D633–D639 (2018).
Fernandez-Pozo, N. et al. The Sol Genomics Network (SGN)-from genotype to phenotype to breeding. Nucleic Acids Res. 43, D1036–D1041 (2015).
Foerster, H. et al. SolCyc: a database hub at the Sol Genomics Network (SGN) for the manual curation of metabolic networks in Solanum and Nicotiana specific databases. Database 2018, bay035 (2018).
Toronto International Data Release Workshop Authors et al.Prepublication data sharing. Nature 461, 168–170 (2009).
Zheng, Y. et al. Cucurbit Genomics Database (CuGenDB): a central portal for comparative and functional genomics of cucurbit crops. Nucleic Acids Res. 47, D1128–D1136 (2019).
Cheng, F. et al. BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol. 11, 136 (2011).
Wang, X. et al. HMOD: an omics database for herbal medicine plants. Mol. Plant 11, 757–759 (2018).
Lee, T. H., Tang, H. B., Wang, X. Y. & Paterson, A. H. PGDD: a database of gene and genome duplication in plants. Nucleic Acids Res. 41, D1152–D1158 (2013).
Qiao, X. et al. Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 20, 38 (2019).
Tang, H. B. Disentangling a polyploid genome. Nat. Plants 3, 688–689 (2017).
Zhu, T. et al. Sequencing a Juglans regia×J. microcarpa hybrid yields high-quality genome assemblies of parental species. Hortic. Res. 6, 55 (2019).
Wu, G. A. & Gmitter, F. G. Novel assembly strategy cracks open the mysteries of walnut genome evolution. Hortic. Res. 6, 57 (2019).
Smedley, D. et al. The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res. 43, W589–W598 (2015).
Yano, K. et al. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 48, 927–934 (2016).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Li, X. et al. Improved hybrid de novo genome assembly of domesticated apple (Malus x domestica). Gigascience 5, 35 (2016).
Daccord, N. et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 49, 1099–1106 (2017).
Bowers, R. M. et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731 (2017).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31801898), the Natural Science Foundation of Fujian Province, China (Kjd18033A), open funds of the State Key Laboratory of Crop Genetics and Germplasm Enhancement (ZW201909), the State Key Laboratory of Tree Genetics and Breeding (TGB2018004), and the Outstanding Youth Program of Fujian Agriculture and Forestry University.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, F., Song, Y., Li, X. et al. Genome sequences of horticultural plants: past, present, and future. Hortic Res 6, 112 (2019). https://doi.org/10.1038/s41438-019-0195-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-019-0195-6
This article is cited by
-
Regulation of plant epigenetic memory in response to cold and heat stress: towards climate resilient agriculture
Functional & Integrative Genomics (2023)
-
History, evolution and domestication of garlic: a review
Plant Systematics and Evolution (2023)
-
Developing future heat-resilient vegetable crops
Functional & Integrative Genomics (2023)
-
Recent advances in proteomics and metabolomics in plants
Molecular Horticulture (2022)
-
The application of CRISPR/Cas technologies to Brassica crops: current progress and future perspectives
aBIOTECH (2022)
