
Abstract
Cauliflower is an important variety of Brassica oleracea and is planted worldwide. Here, the high-quality genome sequence of cauliflower was reported. The assembled cauliflower genome was 584.60 Mb in size, with a contig N50 of 2.11 Mb, and contained 47,772 genes; 56.65% of the genome was composed of repetitive sequences. Among these sequences, long terminal repeats (LTRs) were the most abundant (32.71% of the genome), followed by transposable elements (TEs) (12.62%). Comparative genomic analysis confirmed that after an ancient paleohexaploidy (γ) event, cauliflower underwent two whole-genome duplication (WGD) events shared with Arabidopsis and an additional whole-genome triplication (WGT) event shared with other Brassica species. The present cultivated cauliflower diverged from the ancestral B. oleracea species ~3.0 million years ago (Mya). The speciation of cauliflower (~2.0 Mya) was later than that of B. oleracea L. var. capitata (approximately 2.6 Mya) and other Brassica species (over 2.0 Mya). Chromosome no. 03 of cauliflower shared the most syntenic blocks with the A, B, and C genomes of Brassica species and its eight other chromosomes, implying that chromosome no. 03 might be the most ancient one in the cauliflower genome, which was consistent with the chromosome being inherited from the common ancestor of Brassica species. In addition, 2,718 specific genes, 228 expanded genes, 2 contracted genes, and 1,065 positively selected genes in cauliflower were identified and functionally annotated. These findings provide new insights into the genomic diversity of Brassica species and serve as a valuable reference for molecular breeding of cauliflower.
Similar content being viewed by others


Introduction
The genus Brassica contains three basic genomes (A, B and C) that form three diploid species, namely, Brassica rapa (AA genome), Brassica nigra (BB genome), and Brassica oleracea (CC genome), which further hybridize to give rise to three allopolyploid species, namely, Brassica napus (AACC genome), Brassica juncea (AABB genome), and Brassica carinata (BBCC genome), as described by the triangle of U model1. These Brassica species encompass many important vegetable and oilseed crops, such as Chinese cabbage, turnip, cabbage, broccoli, cauliflower, and oilseed rape. Among them, cauliflower (B. oleracea L. var. botrytis, CC genome) is an important variety of B. oleracea that differs from most Brassica species in its formation of a specialized organ called the curd during floral development2. Curds are composed of many indeterminate inflorescences and shortened inflorescence branches3,4 and are the primary edible organs of cauliflower, with abundant nutrient materials, such as sulforaphane5. Sulforaphane is regarded as one of the most powerful natural bioactive substances in preventing and/or defending against cancers6,7,8,9,10. In the past decade, the genomes of several Brassica species, including the B. rapa cultivar Chiifu11; B. oleracea cultivars 02-1212, TO1000DH13, and HDEM14; B. nigra cultivar YZ1215115; B. napus cultivars Darmor-bzh16, ZS1117, and Tapidor18; and B. juncea cultivar T84-6615, have been explored. These studies provided insights into the evolution and phenotypic diversification of Brassica species. However, our knowledge of genome variation and agriculturally important trait formation in cauliflower, one of the most important vegetable crops, is still lacking. Here, the genome of cauliflower was sequenced by PacBio and Illumina sequencing technologies to further understand the evolution of Brassica species, especially the genetic variation in the C genome of B. oleracea species, and reveal the formation of extreme morphological characteristics, namely, the enlarged inflorescences (curds).
Materials and methods
Plant materials
An advanced-generation inbred line of B. oleracea L. var. botrytis (C-8) was selected for whole-genome sequencing, which is widely used as a parental line for breeding due to its excellent agronomic traits. Ten-day-old seedlings of C-8 were harvested and stored at −80 °C prior to DNA extraction.
Genome sequencing
Genomic DNA was extracted from the 10-day-old seedlings of C-8 by using a DNA Secure Plant Kit (TIANGEN, China) and broken into random fragments. DNA sequencing libraries were constructed in accordance with the standard Illumina library preparation protocols. Paired-end libraries with insert sizes of 350 bp were constructed according to the manufacturer’s instructions (Illumina, CA, USA). All of the constructed libraries were sequenced on an Illumina HiSeq X Ten. At least 10 μg of sheared DNA is required to construct PacBio libraries. SMRTbell template preparation involved DNA concentration, damage repair, end repair, hairpin adapter ligation, and template purification. SMRTbell libraries with an insert size of 20 kb were constructed and then sequenced on a PacBio Sequel platform (Pacific Biosciences, CA, USA) by using P6 polymerase/C4 chemistry in accordance with the manufacturer’s procedure (Pacific Biosciences, CA, USA).
Estimation of genome size
Genome size was estimated by K-mer distribution analysis. Approximately 45 Gb of high-quality paired-end reads (350 bp) was generated and used to determine the abundance of 17-nt K-mers. The distribution of 17-nt K-mers depends on the characteristics of the genome and follows a Poisson distribution.
Genome assembly and quality evaluation
The de novo assembly of PacBio single-molecule long reads from Single Molecule Real Time (SMRT) sequencing was performed by using FALCON19 (https://github.com/PacificBiosciences/FALCON/). The 60 subreads with the longest coverage were first selected as seed reads for error correction to obtain enough corrected reads. Then, the error-corrected reads were aligned to one another and assembled into genomic contigs by using FALCON with the following parameters: length_cutoff_ pr = 5,000, max_diff = 120, and max_cov = 130. The draft assembly was polished using the Quiver algorithm. Subsequently, Pilon20 was used to perform error correction of p-contigs with the short paired-end reads generated from an Illumina HiSeq platform. The draft assembly was evaluated by mapping the high-quality reads from short-insert-size libraries to the contigs by using BWA-MEM21. The distribution of the sequencing depth at each position was calculated with SAMtools22 to assess the completeness of the assembled genome. The GC content distribution of the assembled genome was examined to analyze the nucleotide distribution, assess the randomness of sequencing, and check for possible sample contamination. The Core Eukaryotic Genes Mapping Approach (CEGMA)23 pipeline was used to assess the completeness of the genome assembly, which was further assessed by Benchmarking Universal Single-Copy Orthologs (BUSCO)24 analysis.
Transcriptome sequencing
Four tissues of C-8 (leaf, curd, flower and root) were collected for RNA-seq analysis, and four sequencing libraries were constructed from these tissues using an Illumina standard mRNA-seq prep kit.
Genome annotation
TEs and other repetitive sequences in the C-8 genome were searched by combining de novo- and homology-based approaches. For the de novo-based approach, RepeatModeler25, LTR_FINDER26, and RepeatScout27 were used to build a de novo repeat library. For the homology-based approach, the Repbase TE library and the TE protein database were searched against the cauliflower genome by utilizing RepeatMasker (version 3.3.0)28 and RepeatProteinMask, respectively. Tandem repeats were detected in the genome by using Tandem Repeats Finder software29.
Homology-based prediction and de novo-based prediction combined with transcriptome-based prediction were conducted to predict the protein-coding genes in the cauliflower genome. Homologous proteins from six plant genomes (Arabidopsis thaliana, Arabidopsis lyrata, B. oleracea, B. rapa, Capsella rubella, and Raphanus sativus) were downloaded from the Ensembl and NCBI databases. The protein sequences from these genomes were aligned to the cauliflower genome assembly by using TBLASTN30 with an E-value cutoff of 1e-05. BLAST hits were conjoined by Solar software31. GeneWise32 was used to predict the exact gene structure of the corresponding genomic regions for each BLAST hit (Homology-set). For transcriptome-based prediction, RNA-seq data were mapped to the assembly by using TopHat (version 2.0.8)33, and Cufflinks (version 2.1.1)34 (http://cufflinks.cbcb.umd.edu/) was then used to assemble the transcripts into gene models (Cufflinks-set). In addition, the RNA-seq data were assembled by Trinity35 to create several pseudo-ESTs. These pseudo-ESTs were also mapped to the assembly, and gene models were predicted by PASA36. This gene set was denoted PASA-T-set (PASA Trinity set) and used to train ab initio gene prediction programs. Five ab initio gene prediction programs, namely, Augustus (version 2.5.5)37, GENSCAN (version 1.0)38, GlimmerHMM (version 3.0.1)39, geneid40, and SNAP41, were used to predict the coding regions in the repeat-masked genome. Gene model evidence from the Homology-set, Cufflinks-set, PASA-T-set and ab initio programs were combined by EVidenceModeler (EVM)42 into a nonredundant set of gene structures.
Functional annotation of the protein-coding genes was achieved using BLASTP (E-value of 1e-05)43 against two integrated protein sequence databases: SwissProt and NR. Protein domains were annotated by searching against the InterPro (V32.0)44 and Pfam (V27.0)45 databases, using InterProScan (V4.8)46 and HMMER (V3.1)47, respectively. The Gene Ontology (GO) terms48 for each gene were obtained from the corresponding InterPro or Pfam entry. The pathways in which the genes might be involved were assigned by BLAST against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (release 53)49, with an E-value cutoff of 1e-05.
The tRNA genes were identified by tRNAscan-SE software50. The rRNA fragments were predicted by aligning the rRNA sequences by using BLASTN at an E-value of 1e-10. The miRNAs and snRNAs were predicted by INFERNAL software51 against the Rfam database (release 9.1)52.
Gene family construction
The protein sequences from cauliflower and other sequenced plant genomes with representatives from A. thaliana, A. lyrata, Aethionema arabicum, B. juncea (AABB genome), B. napus Darmor-bzh (AACC genome), B. napus ZS11 (AACC genome), B. napus Tapidor (AACC genome), B. nigra (BB genome), B. oleracea L. var. capitata (CC genome), B. oleracea TO1000DH (CC genome), B. rapa (AA genome), Cardamine hirsuta, C. rubella, Eutrema salsugineum, Sisymbrium irio and Schrenkiella parvula were used for gene family clustering. The gene set of each species was filtered as follows: first, when multiple transcripts were present in one gene, only the longest transcript in the coding region was obtained for further analysis; second, the genes encoding proteins of fewer than 30 amino acids were filtered out. Then, the similarity relation between the protein sequences of all of the species was obtained through BLASTP with an E-value of 1e-05. All of the protein datasets of the representative plant species were clustered into paralogs and orthologs by using the program OrthoMCL (http://orthomcl.org/orthomcl/)53 with an inflation parameter of 1.5. The three members of Brassica with a C genome, namely, cauliflower, B. oleracea L. var. capitata and B. oleracea TO1000DH, were further clustered and subjected to Venn diagram analysis to explore the species-specific genes in cauliflower. The cauliflower-specific genes were then subjected to GO and KEGG functional annotation.
Phylogenetic tree and divergence time estimation
The single-copy genes in the representative plant genomes were aligned by MUSCLE (http://www.drive5.com/muscle/)54, and the alignment results were combined to create a super-alignment matrix. A phylogenetic tree comprising cauliflower and other representative plant species was constructed by using RAxML (http://sco.h-its.org/exelixis/web/software/raxml/index.html) with the maximum likelihood method, and the number of bootstraps was 1,00055. A. arabicum was designated as the outgroup of the phylogenetic tree. The MCMCTree program of PAML5 (http://abacus.gene.ucl.ac.uk/software/paml.html) was applied to infer the divergence time based on the constructed phylogenetic tree, employing the following parameters: burn-in = 5,000,000, sample number = 1,000,000, and sample frequency = 5056. The calibration time of divergence of these plant species was obtained from the TimeTree database (http://www.time.org/)57.
Detection of polyploidization events
The protein sequences from cauliflower, B. nigra, B. napus Darmor-bzh, B. oleracea TO1000DH, and B. rapa were searched against themselves by using BLASTP (E-value < 1e-05) to identify syntenic blocks. The results were also subjected to MCScanX58 to determine the syntenic blocks. At least five genes were required to indicate synteny. Subsequently, the protein sequence alignments were converted into a CDS file, and 4DTv values were calculated on the basis of CDS alignments accompanying the correction of the HKY model.
Gene family expansion and contraction analysis
The expansion and contraction of the gene families were analyzed by comparing the cluster size differences between the ancestor and each species by using the CAFÉ program59. A random birth and death model was used to study the changes in gene families along each lineage of the phylogenetic tree. A probabilistic graphical model (PGM) was introduced to calculate the probability of transitions in gene family size from parent to child nodes in the phylogeny. Using conditional likelihoods as the test statistics, the corresponding p-value in each lineage was calculated, and a p-value of 0.05 was used to identify the families that were significantly expanded or contracted. The expanded and contracted genes were then subjected to GO and KEGG functional annotation.
Positively selected gene analysis
The single-copy genes from cauliflower and two other relative species were aligned using MUSCLE54 to identify the positively selected genes in cauliflower compared with other Brassica species with the C genome. Likelihood ratio tests based on the branch-site models of PAML56 were conducted to detect positive selection, with cauliflower as a foreground branch. p values were computed using the χ2 statistic and corrected for multiple testing by the false discovery rate (FDR) method. The positively selected genes were subjected to GO and KEGG functional annotation.
Results
Genome sequencing and assembly
The cauliflower genome estimated by K-mer analysis was 603.04 Mb. A total of 69.06 Gb of high-quality PacBio long reads (114.52X coverage of the genome) and 45.99 Gb of Illumina clean reads (76.26X coverage of the genome) were generated, resulting in approximately 190.78-fold coverage of the cauliflower genome. All of these reads were further assembled into 584.60 Mb, consisting of 1,484 contigs with a contig N50 of 2.11 Mb and a longest contig of 9.81 Mb, which represented 96.94% of the cauliflower genome (Table 1). The high-quality reads from the short-insert paired-end libraries were mapped to the contigs by BWA-MEM to evaluate the quality of the assembled genome. Approximately 99.43% of the reads could be mapped to the assembly, which covered 99.15% of the assembled sequence (Table S1). The CEGMA and BUSCO pipelines were used to further assess the completeness of the genome assembly. The CEGMA results confirmed the homologs for 96.77% of the core eukaryotic genes in the assembly (Table S2). BUSCO analysis revealed that 97.2% of the genes in the cauliflower genome were conserved. These results verified the high quality of the presently generated cauliflower genome assembly (Table S3). The raw genome and transcriptome sequencing data are available from the NCBI under the project ID PRJNA516731.
Genome annotation
The combined de novo-, homology-, and transcriptome-based predictions revealed that 108.40 Mb of the 584.60-Mb assembled genome (18.54% of the genome) was annotated to code 47,772 genes, with 4.78 exons per gene on average. The average transcript length per gene was 2,035 bp, with average exon and intron lengths of 242 and 260 bp per transcript, respectively. Among the 47,772 predicted genes, 46,628 genes (97.60% of the total genes) could be annotated in at least one functional protein database. The de novo-based and homology-based approaches were also used to search for and predict repetitive sequences. A total of 56.65% of the assembled cauliflower genome was composed of repetitive sequences (Table 1). Among these repetitive elements, LTRs were the most abundant, accounting for 32.71% of the genome, followed by TEs (12.62%), tandem repeats (9.34%), LINEs (5.20%), simple repeats (2.89), and SINEs (0.06%) (Table S4). In addition, 0.23% of the assembled genome was annotated as noncoding RNAs, which included 1,828 miRNAs, 1,408 tRNAs, 2,420 rRNAs, and 2,450 snRNAs (Table S5).
Evolution of the cauliflower genome
The genome sequences of the representative plant species were collected and subjected to comparative genomic analysis with cauliflower to reveal the genome evolution and divergence of cauliflower. The clustering results revealed 64,592 gene families in these plant species, and among them, 3,109 gene families were common. Furthermore, 434 of the 3,109 common gene families contained one copy in each plant species (Fig. 1a). These 434 single-copy orthologous genes were used to construct the phylogenetic tree. The results confirmed that the A, B, and C genomes from different Brassica species were classified into three independent branches, consistent with the results described in the triangle of U model. Brassica species with A and C genomes displayed a shorter genetic distance than the species with the B genome. In the three Brassica species with the C genome, cauliflower and B. oleracea TO1000DH were clustered together on a branch (Figure S1). Furthermore, the molecular clock of these plant genomes was calculated. The calibration times of divergence were 3.2–7.0 Mya between A. lyrata and A. thaliana, 9.1–11.3 Mya between A. thaliana and C. rubella, 12.3–20.6 Mya between C. rubella and C. hirsuta, and 18.1–26.3 Mya between S. parvula and S. irio (Fig. 2). The data further confirmed that the Brassica ancestor diverged from S. irio approximately 16.3–25.0 Mya. In Brassica species, the diploid species B. nigra with the B genome diverged from the A and C genomes approximately 9.1–13.4 Mya. Then, the other two diploid species, namely, B. oleracea (CC genome) and B. rapa (AA genome), diverged ~8.5 Mya. Furthermore, the A genome of diploid B. rapa diverged into the A subgenome of allotetraploid B. juncea 2.3–4.1 Mya and B. napus 2.8–4.8 Mya. The three subgenomes of B. napus diverged 2.4–4.0 Mya. The C genome of the B. oleracea ancestral species diverged into the C subgenome of allotetraploid B. napus, uncultivated B. oleracea TO1000DH (wild cabbage) and two varieties, specifically, cauliflower and B. oleracea L. var. capitata, approximately 3.0 Mya. B. oleracea TO1000DH diverged from the variety B. oleracea L. var. capitata approximately 2.6 Mya, whereas the divergence time between cauliflower and B. oleracea TO1000DH was ~2.0 Mya (Fig. 2).

a Distribution of single-copy orthologs (pink), multiple-copy orthologs (orange), unique genes (brown), and other unclassified genes (green) in cauliflower and other representative plant species. b Venn diagram of gene families in cauliflower and two other Brassica species with the C genome

The nodes represent the divergence time from present (million years ago, Mya). The blue number in the brackets indicates the confidence interval of the divergence time. a–c indicate the A, B and C genomes of Brassica species, respectively
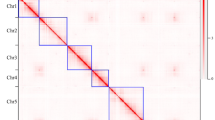
Synteny analysis was conducted in cauliflower and four other representative Brassica species, namely, B. nigra, B. napus Darmor-bzh, B. oleracea TO1000DH, and B. rapa, to further understand the genome evolution of cauliflower. The results indicated that 607 syntenic blocks, which involved 31,380 genes, were present between cauliflower and B. oleracea TO1000DH (Figure 3, Table S6 and S7). Among the 607 syntenic blocks, 103 were distributed on chromosome no. 03 of cauliflower, representing the most syntenic blocks on the 9 chromosomes, followed by 79 on chromosome no. 07; 73 on chromosome no. 04; 71 on chromosome no. 08; 61 on chromosome no. 09; 59 on chromosome no. 02; 58 on chromosome no. 06; 55 on chromosome no. 05; and 48 on chromosome no. 01. A total of 777 syntenic blocks were detected between cauliflower and B. nigra, which were composed of 34,361 genes (Figure 3, Table S8 and S9). Similarly, chromosome no. 03 of cauliflower shared the most syntenic blocks (122) with the eight chromosomes of B. nigra, followed by chromosome nos. 04 and 07. A total of 761 syntenic blocks existed between cauliflower and B. rapa. Among them, 126 were distributed on chromosome no. 03 of cauliflower, also representing the most syntenic blocks on the 9 chromosomes. These combined 761 syntenic blocks were composed of 39,449 genes (Figure 3, Table S10, S11). These results indicated that chromosome no. 03 of cauliflower shared the most syntenic blocks with the A and B genomes of Brassica species and the C genome of B. oleracea. Moreover, synteny analysis of the 9 chromosomes of cauliflower confirmed that chromosome no. 03 shared the most syntenic blocks with the 8 other chromosomes (Fig. 4, Table S12 and S13). These findings suggested that chromosome no. 03 is the most ancient one in the cauliflower genome, which was inherited from the common ancestor of Brassica species.


Chrom. 01, 02, 03, 04, 05, 06, 07, 08, and 09 indicate the nine assembled chromosomes of cauliflower
According to the syntenic blocks detected in cauliflower and four other Brassica species, 4DTv values were estimated. The data revealed three peaks at approximately 0.15, 0.33, and 0.6 in cauliflower and four other Brassica species, whereas only two peaks at approximately 0.6 and 0.33 were detected in Arabidopsis. The peaks at approximately 0.6 and 0.33 revealed two WGD events (β and then α) shared with Arabidopsis after the ancient γ event in cauliflower and other Brassica species. The other peak at ~0.15 indicated an additional WGT event shared among Brassica species (Fig. 5). Moreover, the 4DTv analysis confirmed that the cauliflower genome first diverged from that of B. nigra (BB genome) and then diverged from that of B. rapa (AA genome). The genomes of cauliflower and B. oleracea TO1000DH and the C subgenome of B. napus Darmor-bzh arose recently and did not exhibit significant divergence compared with the A and B genomes of Brassica species. This finding was consistent with the phylogenetic analysis of Brassica and other representative Cruciferae species (Fig. 5).

The x axis indicates the 4Dtv distance. The y axis indicates the percentage of gene pairs. (C) shows the C subgenome of B. napus Darmor-bzh
Specific gene families in cauliflower
Comparative genomic analysis was conducted to determine the specific genes only present in cauliflower. In three Brassica species with the C genome, namely, cauliflower, B. oleracea L. var. capitata and B. oleracea TO1000DH, 20,223 gene families were common. Compared with those in B. oleracea L. var. capitata and B. oleracea TO1000DH, 2,116 gene families, including 2,718 genes, were cauliflower specific (Fig. 1b, Table S14). Functional annotation results confirmed that these genes were significantly enriched in the GO terms involved in respiratory electron transport chain, ribosome, translation, ribonucleoprotein complex, structural constituent of ribosome, RNA–DNA hybrid ribonuclease activity, tryptophanyl-tRNA aminoacylation, intracellular organelle, structural molecule activity, endopeptidase inhibitor activity, and tryptophan-tRNA ligase activity (Table S15). KEGG analysis indicated that these specific genes were mainly involved in metabolic pathways such as mismatch repair, DNA replication, homologous recombination, nucleotide excision repair, oxidative phosphorylation, beta-alanine metabolism, benzoxazinoid biosynthesis and biosynthesis of siderophore-group nonribosomal peptides (Table S16). These data indicated that the cauliflower-specific gene families were mainly involved in the biosynthesis, metabolism, and modification of proteins and nucleic acids.
Expansion and contraction of gene families in cauliflower
The expansion and contraction of the gene families in cauliflower were explored by comparing them with the gene families in B. oleracea L. var. capitata and B. oleracea TO1000DH. The results indicated that 6 gene families, including 228 genes, were significantly expanded in cauliflower, and these genes were mainly involved in peptidyl-prolyl cis-trans isomerase, armadillo repeat-containing kinesin, and senescence-associated protein (Table S17). Functional categories indicated that these expanded genes were significantly enriched in protein folding and zinc ion binding. In contrast, only 2 genes significantly contracted in cauliflower: UDP-glycosyltransferase and elongation factor 1-alpha 4. These contracted genes were significantly enriched in zeatin biosynthesis.
Positively selected genes in cauliflower
The Ka/Ks of the single-copy genes was evaluated by using cauliflower as a foreground branch and two other related Brassica species (B. oleracea L. var. capitata and B. oleracea TO1000DH) as a background branch to identify the positively selected genes in cauliflower. A total of 1,065 candidate genes in cauliflower underwent positive selection (P < 0.05; Table S18). Most of them were enriched in the GO terms involved in nucleic acid binding, cellular nitrogen compound metabolism, organic cyclic compound metabolism, heterocycle metabolism, and nucleobase-containing compound metabolism (Table S19).
Discussion
Cauliflower is a variety of B. oleracea and an important vegetable crop. B. oleracea also includes other vegetable crops, such as cabbage, broccoli, kale, brussels sprouts, collard greens, savoy, kohlrabi, and gai lan. These varieties display very high phenotypic diversity, even though they originated from the same ancestor. Cauliflower and broccoli exhibit extremely enlarged inflorescence meristems and/or flower buds, which form the major edible organ called the curd. Cabbage and brussels sprouts display extremely enlarged terminal leaf buds and lateral leaf buds, respectively. The leaves of kale and collard greens are obviously different from those of uncultivated wild cabbage, whereas kohlrabi has a dramatically enlarged stem, which is its edible organ. With the rapid development of high-throughput sequencing technologies, whole-genome sequencing can now be conducted to reveal the phenotypic diversity and evolution of nonmodel plant species. In Brassica species, the genomes of at least nine species or varieties, including one cultivar from B. rapa with the AA genome11, one cultivar from B. nigra with the BB genome15, three cultivars from B. oleracea with the CC genome12,13,14, three cultivars from B. napus with the AACC genome16,17,18, and one cultivar from B. juncea with the AABB genome15, have been sequenced. In the present study, the genome of another important cultivar from B. oleracea, cauliflower, was uncovered. A 584.60-Mb cauliflower genome assembly with 47,772 genes was reported. The repetitive sequences comprised 56.65% of the genome. The cauliflower genome was larger than the A genome of B. rapa (238.32 Mb)11, the B genome of B. nigra (396.9 Mb)15, and the A or B subgenome of B. napus16,17,18 and B. juncea15. The data confirmed that the cauliflower genome was also larger than that of B. oleracea cultivars 02–12 (539.91 Mb)12, TO1000DH (488.6 Mb)13, and HDEM (545.2 Mb)14 and the C subgenome of B. napus16,17,18. However, the number of genes in cauliflower was similar to or less than that in Brassica species with the C genome. These findings indicated that a high-quality cauliflower genome was generated and suggested that repetitive sequences and other noncoding sequences were more abundant in cauliflower than in other B. oleracea species. The present genomic data provide valuable references for understanding the formation and regulation of important agronomic traits in cauliflower and related species.
Cauliflower was domesticated from wild B. oleracea, which originally grew in the coastal Mediterranean. According to historical records, cauliflower was first introduced to France from Genoa, Italy, in the 16th century, but it did not commonly appear on grand tables until the time of Louis XIV. In the 18th century, cauliflower was introduced to India, and it was introduced to southern China in the middle of the 19th century. Currently, China is one of the countries that cultivates and consumes the most cauliflower. However, the origin and evolution of cauliflower in the genus Brassica is still ambiguous. Nevertheless, genomic data from other Brassica species have provided significant insight into the evolutionary relationships of plant species in the genus Brassica as well as in the whole Cruciferae. The Brassica and Arabidopsis lineages diverged approximately 20–35 Mya60,61. B. rapa and B. oleracea diverged from the A. thaliana lineage 5–911 and 4.6 Mya12, respectively. Good estimates indicated the separation of B. oleracea from B. rapa approximately 4 Mya, and B. nigra separated from the A/C lineage 6–14.6 Mya62,63. The divergence time between B. napus and its progenitors was ~7500 years ago or less, based on different methods of estimation16,17. Consistent with previous investigations, the present cauliflower genomic data and phylogenetic analysis confirmed that the Brassica species with the B genome, such as B. nigra, are more ancient than those of Brassica species with the A or C genome. The Brassica species with the A genome and those with the B genome diverged approximately 8.5 Mya. The Brassica species with the A genome further diverged ~3.8 Mya, whereas those with the C genome diverged ~3.0 Mya. These results implied that the Brassica species with the A genome appeared earlier than those with the C genome. The C genome of the B. oleracea ancestral species diverged into the C subgenome of allotetraploid B. napus and the C genome of diploid varieties, including B. oleracea L. var. capitata, B. oleracea TO1000DH, and cauliflower, implying that the C subgenome in B. napus originated from the genome of the ancestral diploid B. oleracea species, while it was different from the genome of the modern diploid B. oleracea species. The present results also confirmed that B. oleracea L. var. capitata was generated earlier than cauliflower. The cauliflower genome was likely generated ~2.0 Mya, making cauliflower the youngest known variety of B. oleracea as well as in the genus Brassica.
Except for the specialized organ, the curd, the phenotypes of cauliflower are similar to those of closely related species in the genus Brassica, such as uncultivated B. oleracea TO1000DH and B. oleracea L. var. capitata. Especially in the seedling phase, distinguishing one species from another is difficult. Consequently, identifying the differential genes between cauliflower and closely related species is important for uncovering the phenotypic specificity of cauliflower. Here, comparative genomic analysis of cauliflower and two other B. oleracea species, namely, B. oleracea TO1000DH and B. oleracea L. var. capitata, confirmed that 2,116 gene families were cauliflower specific, while only six gene families and two gene families were significantly expanded and contracted in cauliflower, respectively. In addition, compared with the genes in B. oleracea TO1000DH and B. oleracea L. var. capitata, 1,065 positively selected genes were identified in cauliflower. These specific, positively selected, expanded, and contracted genes provided valuable insight into the formation of phenotypic characteristics and evolution of cauliflower. For example, several GO terms or metabolic pathways targeted by the cauliflower-specific gene families displayed significant enrichment in tryptophanyl-tRNA aminoacylation and tryptophan-tRNA ligase activity. Tryptophan is an important precursor of auxin biosynthesis64,65. The only two contracted gene families compared with B. oleracea TO1000DH and B. oleracea L. var. capitata are both involved in zeatin biosynthesis, which plays crucial roles in cytokinin biosynthesis66,67]. These results indicated that novel genes involved in auxin biosynthesis might have been generated during the evolution of cauliflower. However, the genes involved in cytokinin biosynthesis were contracted. Auxin and cytokinin are two important phytohormones that mainly function in regulating plant growth and development. The adjustment of the homeostasis of auxin and cytokinin in cauliflower relative to that in other B. oleracea species might be associated with the phenotypic characteristics of cauliflower.
In conclusion, the high-quality cauliflower genome was reported. Cauliflower underwent two WGD events and one WGT event after the ancient γ event. Chromosome no. 03 may be the most ancient one in the cauliflower genome, as it was inherited from the common ancestor of Brassica species. Cauliflower may be the youngest known variety in the genus Brassica. Moreover, specific, positively selected, expanded and contracted genes that may be closely associated with the phenotypic characteristics and evolution of cauliflower were identified. These findings provide new insight into the genomic diversity of Brassica species and are valuable for molecular breeding of cauliflower.
References
Nagaharu, U. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilication. Jpn J. Bot. 7, 389–452 (1935).
Li, H. et al. Curd development associated gene (CDAG1) in cauliflower (Brassica oleracea L. var. botrytis) could result in enlarged organ size and increased biomass. Plant Sci. 254, 82–94 (2017).
Anthony, R. G., James, P. E. & Jordan, B. R. The cDNA sequence of a cauliflower apetala1/squamosa homolog. Plant Physiol. 108, 441–442 (1995).
Anthony, R. G., James, P. E. & Jordan, B. R. Cauliflower (Brassica oleracea var. botrytis) curd development: the expression of meristem identity genes. J. Exp. Bot. 47, 181–188 (1996).
Cheung, K. L. & Kong, A. N. Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention. AAPS J. 12, 87–97 (2010).
Tarozzi, A. et al. Sulforaphane as a potential protective phytochemical against neurodegenerative diseases. Oxid. Med. Cell. Longev. 2013, 415078 (2013).
Fisher, M. L., Ciavattone, N., Grun, D., Adhikary, G. & Eckert, R. L. Sulforaphane reduces YAP/Np63alpha signaling to reduce cancer stem cell survival and tumor formation. Oncotarget 8, 73407–73418 (2017).
Liu, P. et al. Sulforaphane exerts anti-angiogenesis effects against hepatocellular carcinoma through inhibition of STAT3/HIF-1alpha/VEGF signalling. Sci. Rep. 7, 12651 (2017).
Li, Q. Q. et al. Sulforaphane inhibits cancer stem-like cell properties and cisplatin resistance through miR-214-mediated downregulation of c-MYC in non-small cell lung cancer. Oncotarget 8, 12067–12080 (2017).
Liu, C. M. et al. Sulforaphane targets cancer stemness and tumor initiating properties in oral squamous cell carcinomas via miR-200c induction. J. Formos. Med. Assoc. 116, 41–48 (2017).
Wang, X. et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039 (2011).
Liu, S. et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5, 3930 (2014).
Parkin, I. A. et al. Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 15, R77 (2014).
Belser, C. et al. Chromosome-scale assemblies of plant genomes using nanopore long reads and optical maps. Nat. Plant 4, 879–887 (2018).
Yang, J. H. et al. The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 48, 1225–1232 (2016).
Chalhoub, B. et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953 (2014).
Sun, F. et al. The high-quality genome of Brassica napus cultivar ‘ZS11’ reveals the introgression history in semi-winter morphotype. Plant J. 92, 452 (2017).
Bayer, P. E. et al. Assembly and comparison of two closely related Brassica napus genomes. Plant. Biotechnol. J. 15, 1602–1610 (2017).
Pendleton, M. et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat. Methods 12, 780–786 (2015).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 1303, 3997 (2013).
Li, H. et al. The sequence alignment/map (SAM) format and SAMtools. Transplant. Proc. 19, 1653–1654 (2009).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Smit, A. F. A. & Hubley, R. RepeatModeler Open-1.0. http://www.repeatmasker.org (2008–2015).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, 265–268 (2007).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, 351–358 (2005).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. http://www.repeatmasker.org (1996–2015).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Altschul, S., Gish, W., Miller, W., Myers, E. & Lipman, D. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Yu, X., Zheng, H., Wang, J., Wang, W. & Su, B. Detecting lineage-specific adaptive evolution of brain-expressed genes in human using rhesus macaque as outgroup. Genomics 88, 745–751 (2006).
Birney, E. & Durbin, R. Using GeneWise in the Drosophila annotation experiment. Genome Res. 10, 547–548 (2000).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Grabherr, M. G. et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-seq data. Nat. Biotechnol. 29, 644–652 (2011).
Campbell, M. A., Hass, B. J., Hamilton, J. P., Mount, S. M. & Buell, C. R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genom. 7, 327 (2006).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32, 309–312 (2004).
Aggarwal, G. & Ramaswamy, R. Ab initio gene identification: prokaryote genome annotation with GeneScan and GLIMMER. J. Biosci. 27, 7–14 (2002).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Parra, G., Blanco, E. & Guigó R. GeneID in Drosophila. Genome Res. 10, 511 (2000).
Bromberg, Y. & Rost, B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 35, 3823 (2007).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using evidencemodeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Gish, W. & States, D. J. Identification of protein coding regions by database similarity search. Nat. Genet. 3, 266–272 (1993).
Hunter, S. et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215 (2009).
Finn, R. D. et al. Pfam: the protein families database. Nat. Genet. 42, D222–D230 (2014).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Res. 33, 116–120 (2005).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, 29–37 (2011).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Kanehisa, M. et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205 (2014).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335 (2009).
Griffithsjones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124 (2005).
Li, L., Stoeckert, C. J. Jr. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Hedges, S. B., Dudley, J. & Kumar, S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics 22, 2971–2972 (2006).
Tang, H. et al. Synteny and collinearity in plant genomes. Science 320, 486–488 (2008).
Han, M. V., Thomas, G. W., Lugo-Martinez, J. & Hahn, M. W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997 (2013).
Yang, Y. W., Lai, K. N., Tai, P. Y. & Li, W. H. Rates of nucleotide substitution in angiosperm mitochondrial DNA sequences and dates of divergence between Brassica and other angiosperm lineages. J. Mol. Evol. 48, 597–604 (1999).
Town, C. D. et al. Comparative genomics of Brassica oleracea and Arabidopsis thaliana reveal gene loss, fragmentation, and dispersal after polyploidy. Plant Cell 18, 1348–1359 (2006).
Navabi, Z. K. et al. Conserved microstructure of the Brassica B Genome of Brassica nigra inrelation to homologous regions of Arabidopsis thaliana, B. rapa and B. oleracea. BMC Genom. 14, 250 (2013).
Lysak, M. A., Koch, M. A., Pecinka, A. & Schubert, I. Chromosome triplication found across the tribe Brassiceae. Genome Res. 15, 516–525 (2005).
Bartel, B. Auxin biosynthesis. Ann. Rev. Plant Physiol. Mol. Biol. 48, 51–66 (1997).
Kasahara, H. Current aspects of auxin biosynthesis in plants. Biosci. Biotechnol. Biochem. 80, 34–42 (2016).
Astot, C. et al. An alternative cytokinin biosynthesis pathway. Proc. Natl Acad. Sci. USA 97, 14778–14783 (2000).
Acknowledgements
We are grateful to Dr. Jianhua Zhu, University of Maryland, College Park, USA, for his good advice and critical reading of the manuscript. This work was supported by grants from the National Key Research and Development Program of China (2017YFD0101805), the Modern Agro-Industry Technology Research System, China (CARS-23-A-07), and the Vegetable Modern Agro-Industry Technology Research System, Tianjin (ITTVRS2017004), the Natural Science Foundation of China (No. 31872115). The work reported here was performed in the State Key Laboratory of Vegetable Germplasm Innovation and the Tianjin Key Laboratory of Vegetable Genetics and Breeding, Tianjin, China.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, D., Wang, C., Zhang, X. et al. Draft genome sequence of cauliflower (Brassica oleracea L. var. botrytis) provides new insights into the C genome in Brassica species. Hortic Res 6, 82 (2019). https://doi.org/10.1038/s41438-019-0164-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-019-0164-0
This article is cited by
-
How cauliflower got its curd
Nature Genetics (2024)
-
Genomic analyses reveal the stepwise domestication and genetic mechanism of curd biogenesis in cauliflower
Nature Genetics (2024)
-
Identification of QTLs for resistance to 10 pathotypes of Plasmodiophora brassicae in Brassica oleracea cultivar ECD11 through genotyping-by-sequencing
Theoretical and Applied Genetics (2023)
-
Decoding allelic diversity, transcript variants and transcriptional complexity of CENH3 gene in Brassica oleracea var. botrytis
Protoplasma (2023)
-
Construction of an SNP fingerprinting database and population genetic analysis of 329 cauliflower cultivars
BMC Plant Biology (2022)
