
Abstract
In 2010, a major scientific milestone was achieved for tree fruit crops: publication of the first draft whole genome sequence (WGS) for apple (Malus domestica). This WGS, v1.0, was valuable as the initial reference for sequence information, fine mapping, gene discovery, variant discovery, and tool development. A new, high quality apple WGS, GDDH13 v1.1, was released in 2017 and now serves as the reference genome for apple. Over the past decade, these apple WGSs have had an enormous impact on our understanding of apple biological functioning, trait physiology and inheritance, leading to practical applications for improving this highly valued crop. Causal gene identities for phenotypes of fundamental and practical interest can today be discovered much more rapidly. Genome-wide polymorphisms at high genetic resolution are screened efficiently over hundreds to thousands of individuals with new insights into genetic relationships and pedigrees. High-density genetic maps are constructed efficiently and quantitative trait loci for valuable traits are readily associated with positional candidate genes and/or converted into diagnostic tests for breeders. We understand the species, geographical, and genomic origins of domesticated apple more precisely, as well as its relationship to wild relatives. The WGS has turbo-charged application of these classical research steps to crop improvement and drives innovative methods to achieve more durable, environmentally sound, productive, and consumer-desirable apple production. This review includes examples of basic and practical breakthroughs and challenges in using the apple WGSs. Recommendations for “what’s next” focus on necessary upgrades to the genome sequence data pool, as well as for use of the data, to reach new frontiers in genomics-based scientific understanding of apple.
Similar content being viewed by others

Introduction
In 2010, a major scientific milestone was achieved: publication of the first reference whole genome sequence (WGS) for cultivated apple (Malus domestica). Reported by Velasco and co-authors1, the first apple WGS resulted from the collaborative efforts of scientists from 14 institutions in five countries. Apple was the tenth plant genome to be sequenced, after Arabidopsis, rice, poplar, grape, papaya, sorghum, cucumber, maize, and soybean2. A WGS ideally provides the identity of each nucleotide, in order for each chromosome, in a genome. As this sequence is largely consistent across the nuclei of all living cells of an organism, the WGS fundamentally represents an organism’s internal genetic instructions. “Golden Delicious” was chosen to represent the crop from this genomic perspective because it is a century-old cultivar that remains popular in commercial production across the globe3 and is also highly prominent in the recent ancestry of a large proportion of modern apple cultivars4,5,6,7. Similar to other apple cultivars, “Golden Delicious” is highly heterozygous (3.2 single nucleotide polymorphisms [SNPs] per 1000 bp and π = 0.0032 ± 0.00321. Sequence fragments were compiled in silico into overlapping, contiguous segments (i.e., contigs) then anchored to their most likely positions along the 17 chromosomes of apple. The anchoring used a six-family (n = 720 offspring total) genetic map with 1643 genetic markers, mostly SNPs from the “Golden Delicious” genome itself1. With this approach, contigs spanning 75% of the genome were determined to be correctly oriented, thus leaving 25% of contigs with “uncertain orientation”. This apple WGS covered approximately 81% of the genome, as the average length of assembly was 604 Mb compared to the estimated genome size of 742 Mb1. The N50 for this original WGS was only 16.7 kb (i.e., half the assembly was composed of contigs ≥16.7 kb in length). This WGS was described as a “high-quality draft”—not fully complete, but well worth releasing to the scientific community.
Excitement surrounded publication of the apple WGS: it promised to reveal how apple as a species and a crop arose, to help elucidate how apple trees and organs function, and to provide “a tool to initiate a new era” in apple genetic improvement via breeding. Key basic discoveries reported by Velasco and co-authors1 were: (1) the 17 chromosomes of the apple genome are monophyletically derived from a relatively recent genome-wide duplication of an ancestral 9-chromosome Rosaceae ancestor, giving a particular pattern of chromosomal homologies; (2) the gene pool of cultivated M. domestica was formed primarily from the wild species M. sieversii; (3) the distinctive pome fruit of apple and its close relatives appears to be the result of expansion of a MADS-box fruit-development gene family; and (4) gene families involved in metabolism of sorbitol, the primary transport molecule in Rosaceae for photosynthesis-derived carbohydrates, are also expanded in the apple genome compared to non-Rosaceae genomes. Although it complicated genome assembly, the high heterozygosity of the sequenced cultivar was embraced intentionally for the “practical goal […] to accelerate the breeding of this economically important perennial crop species”1. The authors reported specifically on the genomic positioning of candidate genes for apple production and consumer traits. Allelic differences in the form of SNPs between the two parental homologs of each “Golden Delicious” chromosome were compiled to accelerate cultivar development during breeding. The publication also highlighted the availability of sequences and revealed positions for nearly all genes of apple, to enable genome-wide research of gene functions and their association with traits of value.
A new, very high quality apple WGS has recently succeeded this first apple WGS and now serves as the reference genome for basic and practical scientific advances. A doubled-haploid derivative of “Golden Delicious” known as GDDH13 was sequenced to unprecedented depth with short and long read sequencers and assembled de novo8 in combination with optical maps. The total length of this assembly was 643 Mb, with 42,140 annotated protein-coding genes. The publication investigated the highly repetitive “dark matter” of the genome—transposable elements (TEs), which represented 60% of the genome in the final assembly and provided insights into apple genome evolution8. Just prior to the GDDH13 WGS publication, an improved apple WGS of heterozygous “Golden Delicious” was reported that increased the N50 of contigs from the original WGS by seven-fold to 112 kb9. The GDDH13 N50 was massively higher again, at 5.558 Mb8. At present, when referring to the apple WGS, researchers almost always mean the GDDH13 reference genome.
As promised, the apple WGS has indeed served as a springboard to further scientific advances by the research community. The original paper has been cited almost 1000 times in the eight years since publication, according to Nature Genetics. The following series of vignettes is a non-exhaustive collection describing how the apple WGS (the original as well as the new reference genome, GDDH13) has facilitated fundamental discoveries across the world about M. domestica and served as a resource for practical applications in apple crop improvement. Topics cover both physiological and genetics advances, separately or intertwined. Almost a decade of research has identified challenges and limitations to scientific aspirations in using the apple WGS, some of which have successfully been overcome with the GDDH13 WGS8 or are otherwise being creatively addressed. We highlight opportunities to reach new heights in genomics-based scientific advancement of apple.
Genome-wide SNP marker development: capturing allelic variation
The publication of the first high-quality apple reference genome1 and following updates8 have enabled whole genome investigations for this fruit crop. Reference-based variation discovery approaches compare re-sequencing information to the reference genome assemblies, in order to identify differences between them. Several different approaches exist for this analysis. The first, and conceptually most logical, is high-coverage whole genome sequencing where one or more plants are independently resequenced and compared to the reference through de novo or reference-based assembly. This approach has the advantage of giving potential access to the whole variation (SNPs, insertions, deletions, transversions, copy number variants, etc.) of the resequenced plants, but has the drawback of high sequencing costs (long reads and chromosome scale scaffolding techniques are needed), requires quite complex data analysis, and demands high-performance computing platforms. A simpler approach would be to focus only on SNPs and small insertions/deletions (indels). In this case, sequences (mostly short Illumina reads) can be aligned directly to the reference genome to identify variants. This approach has been followed in many studies both on a whole genome level and by making use of restricted genome libraries such as in genotyping-by-sequencing (GBS).
Three SNP genotyping arrays have been developed for apple. An initial Illumina 8K SNP array was developed from whole-genome resequencing of 27 cultivars at low sequencing coverage10. For the Illumina 20K SNP array, the discovery panel consisted of 14 major founders of European breeding programs at relatively high sequencing coverage11. Most recently, a total of 67 resequenced accessions were studied to build the largest SNP genotyping array yet available for a fruit tree, the Affymetrix Apple480K12. The advantages of these genome-scanning tools include their availability to the whole research community, ability to assay a consistent set of loci each time they are used, and rapid production of results that are relatively easy to interpret for hundreds to thousands of individuals. A drawback is ascertainment bias, because the SNPs included in each array were obtained from discovery panels of particular accessions that were purposely chosen to represent modern breeding germplasm of M. domestica scion cultivars. Therefore, these arrays might not suit some specific research needs where rare or other Malus species-specific variants are important.
The GBS approach, in contrast, does not use a fixed set of SNPs—with corresponding advantages and disadvantages. GBS resources have been used to generate a saturated genetic linkage map of the apple genome13 and for genome-wide association study (GWAS) of fruit quality, harvest date, scab resistance, and leaf shape14,15,16,17. However, GBS generates a sparse genotype matrix due to uneven sequence coverage across individuals screened from the germplasm and across loci. SNP-calling pipelines for GBS designed specifically for highly heterozygous and polyploid species that incorporate haplotype phasing and imputation promise to significantly enhance the utility of GBS18,19,20,21. Aggregation of sequence information across each haploblock22 might be another strategy to improve genome-wide genotyping efficiency. Imputing haplotypes for haploblocks and determining their contribution to QTLs is already possible using dense SNP array data, even in polyploids such as rose23.
Future investigations in this area should: (i) expand the concept of a single genome for apple towards that of the pan-genome concept, whereby the genomic variation of the entire crop is captured, annotated, and made available to the research community, through tools capable of highlighting M. domestica intraspecific sequence differences and associating them with phenotypic effects; and (ii) incorporate wild relatives of apple to enable scouting for the genomic variants responsible for valuable attributes for introgression into apple cultivar breeding programs.
Genetic linkage mapping: increased accuracy for the recombination context of the genome
Genetic linkage maps are employed in marker-trait discovery studies, map-based cloning of genes and the assembly and/or validation of genome sequences, as well as in genetic diversity and relatedness studies that make use of marker haplotypes7,24. A genetic linkage map presents an estimated order and recombination distance of loci along each chromosome composing the genome of a species. Genetic maps constructed to date have mostly resulted from co-segregation analyses of DNA-based markers in full-sib families. Marker ordering can also be based on high-quality reference genome sequences, such as that for peach25. This latter approach can enhance downstream genetic studies by reducing effort required for map construction and by enhancing the quality of the genotypic data, because small regions of double recombination are likely to arise from genotyping errors and spurious genotypes can be easily traced, examined and curated where necessary26. Linkage maps generated de novo might still be useful for validation and improvement of already available whole genome sequences, for ordering de novo assemblies of wild Malus species, and for identifying and validating genomic re-arrangements and large indels.
The original “Golden Delicious” WGS1 did not allow adequate genome-wide ordering, but subsequent research building on it has helped. From 11 to 36% of markers used in genetic linkage map development showed mismatches between genetic and WGS v1.0 physical positions8,27,28,29,30. Nevertheless, physical position information was successfully exploited in designing the 8K, 20K, and 480K apple SNP arrays, where sets of SNP markers were developed for narrow physical bins10,11,12. This design supported the use of haploblocks of aggregated SNPs in genetic analyses, facilitating the development of a new approach for the construction of high quality linkage maps in outbreeding species7,30,31. The most comprehensive linkage map in apple to date was developed in this manner, the iGL map represents more than 3000 meioses derived from 21 full-sib families and 15,417 SNP markers30.
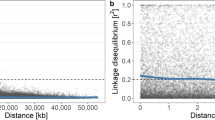
Although the more recent GDDH13 WGS is an improvement over the original WGS8,30, inconsistencies between genetic and physical positions still exist. For example, the gene Mal d 1.06 was mapped genetically to linkage group (LG) 6 by means of a gene-specific sequence characterized amplified region (SCAR) marker, while the marker’s primer sequences were retrieved within a known Mal d 1 gene family cluster on GDDH13 chromosome 1332. In addition, 127 (0.8%) of the 15,417 SNP markers of the iGL map could not be retrieved, suggesting the presence of indels or missing chromosome fragments. Current detailed analyses of LG 1 (Fig. 1) indicate that the iGL map and the GDDH13 v1.1 sequence are highly compatible in most positions. However, the iGL map located two GDDH13 v1.1 regions where the assembly might need further improvement, namely the first 5.8 Mb of the chromosome (where the centromere is located) and the 22.3–28.4 Mb region (Fig. 1), where LG-chromosome and cM-Mb mismatches, unlikely duplications, and un-retrieved iGL markers were noted (Fig. 1). Missing sequence information from the apple WGS was reported where the promoter of MdCPD1 (chromosome 1, 26.2 Mb) could not be retrieved33, and as well as a similar case where cloned MdVQ genes that initially had been identified in the “Golden Delicious” sequence could not be recovered from the GDDH13 v1.1 sequence34. Hence, use of the GDDH13 WGS in marker ordering, as well as for other uses, is not yet straightforward. In the near future, it might be useful to extend the co-linearity between highly curated genetic maps and the GDDH13 WGS to the entire genome and tag suspicious regions in genome browsers.

Haploblock markers of LG 1 plotted for position on the GDDH13 reference WGS compared to that on the iGL genetic linkage map.  = matching order.
= matching order.  = matching after shuffling within genetic bins.
= matching after shuffling within genetic bins.  ,
,  = the two positions of markers with two full hits on chromosome 1 whereas their cluster plots showed them to be true single locus markers; only the former position matched the iGL map (one haploblock with five SNPs jointly representing two continuous stretches of 2.9 kb with 99.8% sequence similarity).
= the two positions of markers with two full hits on chromosome 1 whereas their cluster plots showed them to be true single locus markers; only the former position matched the iGL map (one haploblock with five SNPs jointly representing two continuous stretches of 2.9 kb with 99.8% sequence similarity).  = cM-Mb mismatches that could not be resolved through shuffling on the iGL map and for which calls and hit-qualifications were appropriate; they matched with a 0.8 to 5.5 Mb shift along the chromosome (12 haploblocks with 35 SNPs).
= cM-Mb mismatches that could not be resolved through shuffling on the iGL map and for which calls and hit-qualifications were appropriate; they matched with a 0.8 to 5.5 Mb shift along the chromosome (12 haploblocks with 35 SNPs).  ,
,  = no hit on chromosome 1 but on another chromosome (four haploblocks with seven SNPs) or no hit at all (three haploblocks with four SNPs), respectively; their physical coordinates were estimated based on the nearest matching flanking markers on the iGL map
= no hit on chromosome 1 but on another chromosome (four haploblocks with seven SNPs) or no hit at all (three haploblocks with four SNPs), respectively; their physical coordinates were estimated based on the nearest matching flanking markers on the iGL map
Apple domestication: a story of gene flow
The domestication history of apple started in Central Asia, spreading towards the West in Europe and beyond, assisted by propagation of interesting genotypes by grafting35. Recent investigations using population genetic approaches with microsatellite markers have demonstrated that wild-to-crop gene flow played a major role in the evolutionary history of the cultivated apple36. Reciprocally, crop-to-wild hybridizations have spread alleles from the cultivated gene pool to populations of wild Malus species37,38,39, with possible negative fitness consequences for the wild gene pool39. Such hybridizations, followed by successful establishment of wild beneficial alleles into domesticated populations and vice versa, can occasionally trigger adaptation. For example, adaptive introgression from highland teosintes contributed to maize highland adaptation40. However, so far no study has investigated the adaptive consequences of crop-wild introgressions in apple and their genomic bases. Questions of particular relevance include: What is the genomic architecture of introgressions (number of genes involved, locations, and nature of the genes and their alleles) between the cultivated apple and its wild apple relatives? Have these introgressions been selected upon during apple evolution or domestication, and what form do they take (e.g., amino-acid substitution, gene duplications, gene gains, gene losses)? More generally, are these introgressions adaptive? Resolving the last question, i.e., demonstrating the fitness advantages of the introgressed allele and its associated attribute for the recipient population, would be time-consuming or even infeasible in apple because its long generation time would render field experiments challenging35,41. Fortunately, next generation sequencing technologies can be used to elucidate the first two questions by detecting genomic variation underlying adaptation of wild and cultivated apple populations to their biotic and abiotic environments and fruit-related traits.
Numerous publications involving genome-wide data are accumulating for fruit crop perennials8,42,43,44,45,46,47,48,49,50,51,52,53,54, but they still lag behind those for other crop plant systems41,55,56. In apple, the few studies available have helped to gain initial insights into the genomic basis of apple evolution and domestication8,35,55. For example, a GWAS that included both the cultivated apple and, for the first time, several wild apple species (in particular M. sieversii, M. orientalis Uglitz., and M. sylvestris Mill.) revealed genomic regions determining fruit firmness and flavor and explained how these regions have been affected by selection during domestication57. This study supported a model of apple fruit size evolution comprising two major events, with one occurring prior to domestication and the other during domestication. Similarly, but without the use of genome-wide data, a work by Yao and co-authors58 showed that a microRNA whose expression is associated with fruit size was fixed in cultivated apple and in their wild progenitors with large fruit, indicating that selection for fruit size was initiated before apple domestication. Next generation sequencing technologies have also been used to identify the genes underlying resistance to pathogens, such as blue mold (Penicillium expansum) infection in M. sieversii compared to M. domestica59 and to identify trait loci controlling resistance to this pathogen in M. sieversii60. Genomic analyses therefore suggest that apple differs from annual crop models, such as maize40, for which most key agronomic traits were likely selected after domestication61. However, so far, these genomic studies of apple have not integrated the estimation of the timing of selection associated with such pre-existing or post-domestication adaptation to a new environment (e.g., climate, pathogens, and human) and have not tested whether such genomic regions are the result of crop-wild introgressions following apple domestication.
Wider sampling of the wild apple relatives across their natural Eurasian distributions, in Central Asia, the Caucasus, and Europe (M. sieversii, M. orientalis, and M. sylvestris, respectively), as well as of local M. domestica cultivars from the same regions, and more ancestral species (M. baccata, M. floribunda, M. florentina, M. niedzwetzkyana), will help resolve the timing of adaptation and the role of gene flow in apple evolution and domestication. Indeed, sequential temporal wild-crop introgressions have been previously detected using microsatellite markers from the East (e.g., from M. orientalis) to the West (e.g., from M. sylvestris)35. Development of new population genomic inference methods that allow estimates of the dynamics of gene flow and positive selection between populations62,63,64,65, combined with a comprehensive sampling of both local wild populations and cultivars, now offer clear opportunities to explore the timing, genomic architecture, and regimes of selection acting in the introgressed regions in wild and cultivated apple and will help clarify the role of gene flow in this fascinating perennial model system.
Characterization of germplasm collections: diversity, origins, and worldwide pedigree connections
More than 10,000 apple cultivars have been described worldwide66. Thousands of these cultivars are curated in large national repositories, in addition to those managed by private institutes or associations and active amateur networks, especially in Europe and the U.S. Until the end of twentieth century, characterization of each cultivar was mostly phenotypic, using pomological traits, phenology, fruit sensory quality, etc. Genetic characterization with simple sequence repeat (SSR) markers has been performed intensively in the last 10–15 years, allowing identity checking and designations of synonym groups (containing synonymous cultivars and sports), assessment of the genetic structure at the collection or multi-collection level and preliminary parentage inferences among cultivars (e.g., see ref. 67,68,69).
The apple 20K and 480K SNP arrays11,12, enabled by the apple WGSs1,8, have been widely used to genotype numerous apple cultivars from various, mostly European, germplasm collections. The high-resolution genetic information developed allows unprecedented parentage inferences, revealing a very dense and unsuspected kinship network, with key cultivars at the top of the European pedigree including “Reinette Franche”, “Margil”, and “Alexander”70. Parentage of numerous well-known cultivars such as “White Transparent”, “Ribston Pippin”, and “Braeburn” has been elucidated. Recent extension of the analysis to the wild apple gene pool included resequencing of more than 110 domesticated and wild accessions and resulted in a comprehensive model of apple speciation and domestication along the Silk Road, identifying introgressions from the European M. sylvestris and signatures of selective sweeps underlying loci influencing important fruit quality traits, notably fruit size57.
Numerous short-term and long-term research opportunities beckon. In the short term, evaluating germplasm for allelic variation at breeding-relevant trait loci will help reveal the practical value inherent in collections. Additional outcomes are expected from pedigree reconstruction to infer the European ancestors of numerous heritage U.S. cultivars71 and to better decipher pedigree relationships across Northern and Southern Europe genepools. Calculation of haplotype sharing along and beyond the inferred pedigrees will make it possible to identify and visualize portions of the genome shared (or not) between two or more cultivars, with major implications for fostering crossing and “Breeding by Design” approaches72. Identification of signatures of empirical selection across and beyond the inferred pedigree should be sought to further decipher genomic regions and corresponding genes under selection in the past, with expected insights into the history of apple breeding over previous centuries. Also, effective population sizes in both germplasm and breeding populations, accounting for pedigree information, should be determined, with implications for management of genetic diversity and filling gaps in collections. In the longer term, future research might address the exploration of core and pan-genomes, taking advantage of extensive resequencing of germplasm collections (including wild Malus species), and exploring the putative role of copy number variants (CNVs) in the wide phenotypic variation of apple and related Malus species. The location of genomic regions with haplotypes introgressed from wild Malus species (especially M. sylvestris and M. orientalis) in the domesticated pool should help to assess their impact on adaptive and agronomic value. Another exciting domain is the exploration of epigenetic modifications, taking advantage of bisulfite resequencing or other approaches, especially in very old or widely-dispersed cultivars that have accumulated hundreds of years of modifications, to bring insights into aging and adaptation processes.
Methods for genetic dissection of traits: enhanced Genome-Wide Association Study and Pedigree-based analysis (PBA)
Genetic mapping of trait loci in apple has commonly been performed by interval mapping using bi-parental families. Although the underlying genetic models are simplified in such population structure, genomics-assisted apple improvement will be severely limited if bi-parental families are the primary material for detecting and characterizing apple Mendelian trait loci (MTLs) and quantitative trait loci (QTLs), for three main reasons. Firstly, only the genetic variation segregating within the parents of a cross can be queried, and thus only a small fraction of the apple’s tremendous genetic diversity is examined when only two parents are involved. Secondly, marker-trait associations identified in one family are often not transferable to others, and this lack of transferability is a hindrance to apple breeders eager to exploit marker-trait associations. Finally, the limited number of recombination events captured in bi-parental crosses results in poor mapping resolution, such that gene-level mapping resolution is unlikely and the identification of causal alleles becomes largely guesswork. Some of these limitations can be overcome by using a PBA approach to discover QTLs73,74,75. In this approach, multiple (often smaller) families that are pedigree-connected through immediate and distant progenitors are jointly analyzed. The families are chosen to represent important breeding parents and their alleles, preferably segregating in multiple genetic backgrounds to enable the simultaneous identification and validation of QTLs in a breeding-relevant context76. This approach has been successfully used in multi-institution international projects such as RosBREED77,78 and FruitBreedomics79,80, as well as in national projects81. For QTLs only segregating in certain small families, QTLs might not be detected, their intervals can remain large, and estimates of allele effects can be inaccurate—although these are challenges facing any QTL detection method where allelic contrasts are not well represented in the investigated germplasm.
The release of the apple WGS has been a crucial step in enabling an alternative genetic mapping method, GWAS. GWAS detects genotype–phenotype associations in diverse populations that capture all historical recombination events within the sample82. Thus, GWAS in large, diverse populations overcomes the three limitations of bi-parental linkage mapping outlined above: GWAS captures more diversity, results in improved transferability, and enables higher mapping resolution.
DNA sequence reads generated from next-generation DNA sequencing (NGS) must be aligned to a reference genome before performing GWAS. The effectiveness and accuracy of this alignment relies heavily on the completeness and quality of the reference genome. As described in the previous section on genetic linkage mapping, a substantial proportion of genome-wide markers were assigned to the wrong chromosomes in the apple reference genome described by Velasco and co-workers1. Thus, the current improvements in genome assembly8 should dramatically enhance the success of the very first step in the NGS genotyping process, resulting in higher marker densities, higher quality genotype calls, and ultimately more powerful GWAS.
Genome mis-assembly also makes it difficult to draw conclusions about the genetic architecture of a trait following GWAS. For example, a recent GWAS for apple fruit color identified the well-studied color locus, MYB1, on chromosome 9, but also identified a marker on chromosome 17 significantly associated with color. Can we safely assume then that there are two large-effect loci controlling apple color? The answer in this case is no—only a single locus is involved. The chromosome 17 marker’s position was incorrect, and it actually belonged on its homoeologous chromosome 9 with the other significant markers in and around MYB114. Similarly, incorrect marker order also perturbs widely reported metrics of genomic diversity such as the decay of linkage disequilibrium (LD), which will appear to be more rapid when marker order is muddled in incorrect genome assemblies.
Despite some limitations due to genome assembly errors, GWAS using the original apple reference genome1 has proven an effective alternative to traditional interval mapping approaches. For example, GWAS has identified large-effect loci for firmness, firmness retention, flowering and harvest dates, internal flesh browning and fruit splitting that remained undiscovered, despite numerous interval mapping studies14,17,83,84. Such discoveries are leading to improvements in the efficiency of apple breeding. For example, a firmness marker in the NAC18.1 transcription factor discovered via GWAS14 is currently being applied by Canadian apple breeders.
The release of the apple WGS has reinforced use of the above-mentioned PBA approach by facilitating high-density genome-wide genotyping as well as exploiting haploblocking. The former empowers FlexQTL™’s functionality on estimating genome-wide breeding values, thereby allowing to combine the contributions of statistically distinct QTLs with minor effects associated with genetic bins. The use of haploblocks dramatically reduces computation time with little loss of segregation information24. In this respect, FlexQTLTM based PBA analysis has been ahead of GWAS and genomic prediction approaches when pedigree-connected families are in use.
Physiological roles of apple genes: discovering what the genes do
Understanding gene function in any plant is still a challenge, and this is magnified in a slow-growing perennial such as apple. However, the plummeting cost of sequencing has changed the way researchers can interrogate the physiological roles of apple genes. Reduced costs initially allowed researchers to move from mass sequencing of ESTs85,86 to whole genomes1,8,9 and on to crops, rather than model species, to characterize gene function. The apple WGS now provides the gene and its genomic environment: the promoter, intron-exon structure, flanking sequences, and neighboring genes. Gene expression can be measured by high-throughput sequencing, without the need to choose individual genes for expression studies. Next, rapid resequencing and alignment of diverse genomes can be used to identify allelic differences that lead to the desired physiological effects to enrich for in breeding programs57.
Knowledge of gene function starts with the sequence of the gene, which can be obtained from the WGS with a simple bioinformatic search. Function can be further inferred using knowledge about where the gene is expressed, when and where the corresponding protein is produced, and how the protein interacts with other proteins and compounds to create an effect. Each of these steps requires an increasing amount of research effort. However, the most accurate test is to knock the gene out and/or overexpress the gene to verify its function. This gene perturbation can be performed transiently with a virus-induced gene silencing approach (e.g., see ref. 87) or via a stable transformation (see ref. 88,89,90). The creation of a stable transgenic apple plant was described more than 30 years ago91, but transformation of this crop is still not trivial, restricting the numbers of genes that can be realistically evaluated. The use of model species to rapidly assess apple gene function is one effective method to increase the numbers of genes that can be scrutinized. This pre-functional testing has been demonstrated for the case of genes conferring red coloration92.
With knowledge of the specific genes involved in controlling traits of interest, the possibility opens to use technologies that overcome certain biological limitations of breeding. There are clear breeding targets in apple to generate consumer-attractive apples that are produced in a sustainable, low-impact production line93,94. While transgenic plants are an option in some countries95, breeding programs in more restrictive environments must rely on natural variation to generate new cultivars. New technologies such as CRISPR have been used in apple to create mutations in genes. This application of gene editing does not trigger regulation for genetically modified organisms (GMOs) in the US and in some other countries, but it will be subject to the GMO regulatory process in the EU, even if the mutation is identical to one that has occurred naturally in another variety96. To accommodate this new technology, experts are challenging the European Commission to review the decision of the European Court of Justice in July 201897.
Future research to further understand the physiological roles of apple genes should include the following. Data integration: We have moved from a point where data was hard to generate and was typically over-analyzed to a point where data is easy to generate and often under-analyzed. Large data sets are often treated as a separate entity, with little integration of information from prior publications. There is a massive opportunity to address the way that data is integrated. This involves not only data from apple experiments, but across all plants. Faster gene function analysis: Tools and approaches for higher-throughput gene investigation are needed to further understand how individual apple genes are acting. This will allow more informed choices of genes to focus on as well as alleles to select for in breeding and as CRISPR targets. Focus on the proteins: The way we analyze nucleotides (DNA/RNA) has had a massive revolution in the last 20 years. We need a corresponding revolution in protein-sequencing technologies that are sensitive, high-throughput, and affordable. Once we have this advance, we can begin to dissect the functional biology of apple.
Tree architecture genes and genetics: a growing understanding from the genome
Apple tree architecture is highly organized throughout ontogeny and at different scales. Growth, branching and flowering are the main processes giving rise to plant architecture and are key determinants of productivity. Genetic and genomic study of these traits is hence of great relevance for breeding. Progress in understanding the genetics of architectural traits is limited by the large size and long life of apple trees, which makes it difficult to assess phenotypes on large numbers of individuals. Nevertheless, significant advances have been obtained in genetics studies performed on architectural traits in apple and on the underlying physiological and molecular mechanisms. Early genetic studies underlined the genetic variation in tree forms and architectures of apple98. Heritability was estimated for basic morphological traits such as tree height or trunk diameter99,100 and QTLs mapped for branching habit evaluated qualitatively101 or for global geometric traits such as trunk height and base diameter102. The dissection of architecture into elementary traits led to many QTL mapping studies with bi-parental segregating families, the results of which have suggested complex genetic control103,104.
The apple WGS has supported further advances in understanding the genes and genetic variation controlling apple tree architecture. The WGS has allowed the search for candidate genes in QTL zones, possibly combined with the characterization of mutants or sports that display interesting phenotype105. In apple, the discovery of natural mutants exhibiting a columnar compact growth habit106 is certainly the most well-known, as this habit has been considered suitable for high density orchards. The columnar habit of “Wijcik” was mapped at a locus on LG 10 named Columnar (Co)107. QTLs have been mapped for trunk and branch geometry (length and base diameter) and for sylleptic branching in progenies derived from a columnar parent108,109, most of them colocalized with Co, suggesting pleiotropic effects of Co. The WGS assisted in the fine mapping of Co, through the directed development of genetic markers110,111,112, the identification of BAC clones that jointly covered the Co region113,114,115 and the directed Sanger sequencing of fragments that connected the BAC-derived contigs113,115 or assisted alignment of Illumina whole genome sequencing data to the Co region114 and finally the identification of the causal gene. The columnar phenotype of the dominant allele of the Co locus has been shown to be due to an insertion of a Ty3/Gypsy retrotransposon that likely upregulates a nearby gene encoding a 2OG-Fe(II) oxygenase113,114,115,116.
The WGS has greatly facilitated transcriptomic studies as it has allowed the annotation of differentially expressed genes involved in tree architecture. The columnar mutation was associated with the differential expression of a vast number of genes regulated in the shoot apical meristem117. Another example is the regulation of tree architecture by dwarfing rootstock, which has recently been shown to involve unbalanced carbohydrate allocation and downregulation of auxin influx transporters MdAUX1 and MdLAX2118.
The WGS enables searching for all genes in a given family, as proposed for screening the IGT family genes, that have been assumed to play a crucial role in apple architecture119. Four IGT family members (MdoTAC1a, MdoTAC1b, MdoLAZY1, and MdoLAZY2) have been identified in the apple WGS and characterized in four apple cultivars with contrasting architecture. This study119 revealed mutated sites in promoters and differential expression of these genes in all tissues and organs evaluated, in the four cultivars.
All these studies demonstrate the enormous potential of ongoing genomic investigations to aid in the development of an understanding of the genetic control of modified tree architecture. Results suggest that many of the key genetic pathways are functionally conserved across species120. The logistics of phenotyping large populations of trees with diverse architecture is currently a major bottleneck. If this challenge could be overcome, genomic studies could readily exploit genetically variable multi-family populations composed of related individuals, such as breeding pedigrees or unrelated accessions in germplasm collections. From this perspective, technologies based on terrestrial Lidar and imaging are promising emerging tools121. We can expect that the tremendous genetics advances achieved during the last decade and which are ongoing through the development of high-throughput genotyping, phenotyping, and genomic studies will open new avenues for describing plant architecture and exploring the mechanisms of its genetic and molecular physiological control.
Water and nutrient use efficiency genes and genetics: absorbing new genomics advances
Apple WGSs have helped identify genes and genetic factors involved in the complex and valuable traits of apple tree responses to water and nutrient availability. Water use efficiency (WUE) determines production capacity in regions subject to drought and low water availability and for which advances in knowledge have been made using genomic approaches. WUE is defined by the amount of photosynthesized carbon per units of transpired water and is commonly measured seasonally (units of dry matter seasonal growth per unit of water) or by measuring CO2, O2, and H2O fluxes of tree canopies during short periods122. In combination with phytohormones and root morphology, WUE is thought to be associated with drought tolerance in apple123,124. Phenotypic diversity for physiological and morphological components of WUE was reported in domesticated apple and related wild species and several genes responding to water deficit have been described in apple roots125. Mineral nutrients at the correct concentration play pivotal roles in all physiological functions of apple trees, where a deficiency can lead to fruit disorders such as bitter pit126, while an excess can lead to toxicity disorders such as boron shoot die back127. A perturbation of nutrient-related physiological functions will hence have repercussions on horticultural performance of the whole tree as on apple fruit quality126,128,129,130,131.
Use of the apple WGS v1.0 advanced the genetic dissection of WUE in QTL and gene-based studies. QTLs were identified for proxies of transpiration traits derived from airborne images collected on a “Starkrimson” × “Granny Smith” family that was genotyped with SSR markers132. In another family, three QTLs (on LGs 8, 15, and 16) were identified for carbon isotope discrimination (Δ13C), which is a proxy for seasonal WUE. These QTLs were stable across two years in a “Honeycrisp” × “Qinguan” family, and the LG 8 QTL co-localized with one of the previously mentioned QTLs for transpiration rate133. The WGS allowed the identification of 28 QTL-associated candidate genes and differentially expressed transcription factors between the high and low WUE cultivars “Qinguan” and “Honeycrisp”, respectively134, under drought stress133. Zhou and co-workers135 identified and characterized the MdAGO gene family coding for Argonaute proteins in apple and found them to be induced by drought, salt, cold, and ABA treatment, and MdAGO4.1 was upregulated in the WUE cultivar “Qinguan” during water stress.
Other genes have also been connected to WUE by using apple genomics resources. FK506-binding proteins (FKBPs) play diverse roles in numerous critical processes for plant growth, development, and abiotic stress responses. In apple, 42 putative members of the FKBP gene family were identified across 13 chromosomes by performing BLAST analysis of 23 FKBP proteins of Arabidopsis against predicted apple proteins136. An Arabidopsis ROF1/ROF2-mediated network of 11 other interactive proteins was generated, and their apple homologs were identified on the apple WGS. Subsequent qRT-PCR analysis indicated that, under water-deficit or NaCl treatments, 10 gene pairs were co-expressed and showed uniform upregulation, indicating that genes within this apple MdFKBP62a:MdFKBP65a/b-mediated network have potentially important roles in water-deficit and NaCl-stress signaling136, making them possible targets in breeding for high WUE. RNA binding proteins play important roles in plant responses to biotic and abiotic stresses, including for apple. The YT521-B homology (YTH) domain-containing RNA binding protein (YTP) was first found in Rattus norvegicus and is related to oxygen-deficient stress137. A WGS survey led to the identification of 26 putative YTP gene models in apple138. Next, MhYTP1 and MhYTP2, characterized in M. hupehensis, were implicated in drought response after 6 days and differentially expressed in several plant tissues with and without stress137 and overexpression of this gene enhanced WUE in transgenic plants139,140.
Nutrient uptake and transport efficiency (NUTE) in apple are genetically complex traits specific to each nutrient and involving both root-specific (referring to the rootstock in commercial production conditions) and scion-specific components141,142. NUTE is influenced by a wide range of physiological and morphological mechanisms, such as interaction with soil biota, active, and passive transport, vessel composition and size, root induced hormone concentration124,143, and interaction with pH, as well as management options such as crop load and irrigation144. Classical QTL mapping revealed QTLs influencing scion leaf concentrations for thirteen minerals145. Several of these QTLs have been validated in other experiments and in field applications (G. Fazio, manuscript in preparation). The WGSs and subsequently developed tools such as the 20K SNP array were instrumental to the fine mapping and anchoring of these QTLs as well as to the identification of several candidate genes through gene expression experiments (G. Fazio, manuscript in preparation).
The challenge in the next few years is to develop WGSs that are more relevant and representative of apple germplasm involved root and rootstock traits. More than 750 million trees in the ground worldwide are grafted on “Malling 9” apple rootstock, which is more than any scion cultivar planted, yet except for a few Illumina paired end sequencing efforts no assembled complete rootstock WGS is available. Rootstock cultivars often include species other than M. domestica in their pedigree, such as those derived from “Robusta 5” (Malus × robusta), the source of several valuable rootstock attributes including resistance to fire blight and apple canker146 and improved nutrient uptake. There are opportunities in obtaining genome sequences and developing efficient genome-scanning tools that represent such species to advance rootstock research.
Tree dormancy genes and genetics: awakening well-known candidate genes
Temperate zone fruit trees, including apple, modulate their growth rhythm to adapt to seasonal environmental changes such as temperature and day length. Such trees use bud dormancy to adapt to winter cold. Bud dormancy can be fundamentally defined as the inability of the meristem to resume growth under favorable conditions. Following exposure to a genetically determined specific chilling period, known as chilling requirement (CR) fulfillment, and a subsequent warming period, known as heat requirement (HR) fulfillment, dormant buds can shift to the bud break stage. CR and HR fulfillment associated with winter and spring temperature changes, respectively, are believed to be important for the achievement of uniform flowering in commercial orchards. Global climate change affects chilling accumulation during winter, resulting in the occurrence of abnormal bud dormancy phenology, including morphological bud disorders, bud burst delay, and low bud burst rate. Although current breeding programs mainly focus on improvement of yield, fruit quality, and disease resistance, additional objectives such as climate change adaptation should receive increased attention in the near future. Characterization of genes and genetic mechanisms underlying bud dormancy, CR, HR, bud break, and flowering time is therefore relevant for future apple breeding.
Recent transcriptomic studies using information from the apple WGS has shed light on the possible crucial roles for dormancy regulation in apple of candidate genes for two key transcription factors, DORMANCY-ASSOCIATED MADS-box (DAM) and FLOWERING LOCUS C-like (FLC-like)147,148,149,150,151,152. There is also evidence of metabolite dynamics correlated with many cold-related and dehydration-related gene expression changes during dormancy. Both lipids and raffinose family oligosaccharides accumulated in buds during dormancy, and their biosynthetic and modification pathway-related gene expression changes coincided with dormancy phase transitions153,154. These metabolites might play roles in carbon storage and signal transduction for establishment of dormancy. Strikingly, carbon-limiting conditions and carbon starvation responses underlie bud dormancy in woody and herbaceous species155. Collectively, WGS information has enabled research that has provided new insights into the biochemical pathways underlying dormancy.
The study of flowering time in several apple bi-parental and multi-parental populations revealed a major QTL on LG 9 and secondary QTLs on LGs 8, 12, and 15 depending on the genetic background156,157,158. Candidate genes were identified close to these QTL intervals, especially DAMs and FLC-like homologous genes158. Genomic information from the GDDH13 WGS and the high-density Axiom® Apple480K SNP array enabled a powerful GWAS of flowering time in apple, which confirmed the involvement of LG 9 in the genetic control of flowering time84. Information from the WGS has also facilitated epigenetic studies such as DNA methylation analysis of winter dormancy159. The use of such approaches in the future will also help clarify the possibility that epigenetic memory is coupled with dormancy regulation triggered by environmental signals.
Flowering genes: new knowledge blooming from the genome
Combining the apple WGS with analyses of conservation or divergence of flowering genes offers important clues as to the environmental and physiological factors that influence flowering in apple. Many well-studied flowering genes and gene families in model plant species—including FT, TFL1, SOC1, SPL, LFY, and AP1—are conserved in the apple genome. This observation suggests that the basic genetic wiring controlling flowering, as characterized in other eudicots, can be used as a blueprint for dissecting the genetics of flowering in apple. In contrast, apple has no clear counterparts of many other genes known to have more specialized functions in other plants. For example, apple appears to lack a counterpart of FLC, a MADS-domain transcription factor from Arabidopsis that acts as a major repressor of flowering in the absence of a vernalizing cold period. This absence is consistent with the observation that apple flowers are initiated in mid-summer, independently of cold temperatures.
The relatively recent duplication of the apple genome created two copies of most genes, offering a rich context in which to further explore the diversification of gene function and elaborations on the basic mechanism of flowering in apple. The FT gene is an interesting example. In Arabidopsis, the protein product of the FT gene is produced in the phloem tissues of the leaf and trafficked along the translocation stream to the shoot apical meristem, where it promotes transcription of AP1 and other genes that direct flower formation160,161,162. However, in apple, the duplicated FT genes MdFT1 and MdFT2 are expressed mainly outside the leaf, in distinct patterns, with MdFT1 expressed in the apical meristem during floral induction and MdFT2 in the floral structures and young fruit163,164 (Gottschalk and van Nocker, unpublished results). This situation suggests that FT might mediate promotion of flowering independent of photoperiod, consistent with the apparent day-neutral flowering habit of apple.
The apple WGS will also assist in understanding the effects of plant growth regulators on flowering in apple, potentially leading to development of novel compounds that could increase commercial production efficiency. For example, the phytohormone gibberellic acid (GA) has long been known to repress flowering in apple, in contrast to its well-known promotion of flowering in rosette herbaceous plants165,166. It was recently reported that exogenous GA applied to apple trees early in the growing season was associated with increased expression of MdTFL1-1, one of two copies of the TFL1 gene, late in the growing season, at a time when flowers would be forming167. MdTFL1 can repress flowering when expressed ectopically in Arabidopsis and apple168, and, if MdTFL1 does act as a flowering repressor in apple, then its promotion by GA would provide a simple explanation for the repression of flowering by this phytohormone. Other genes such as TEMPRANILLO (TEM), FLORALTRANSITION AT MERISTEM (FTM1) and SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) could also operate as flowering repressors, as suggested by an approach combining QTL detection and transcriptome analysis169,170.
The reference WGS will also provide insights into the genetic mechanisms underlying juvenility—arguably the most significant challenge for rapid cultivar development171. Indexing the full complement of apple genes, together with the finding that some well-studied mechanisms of phase change in maize and Arabidopsis are conserved in woody perennial plants172, provides a smooth route for developing apple breeding germplasm to enable the rapid introduction of novel phenotypes in response to rapidly changing production environments and market demands. In addition, an improved genomic knowledge of apple will shed light onto the interesting variance in juvenile period length observed among Malus wild and cultivated accessions. The rapid-cycling, wild apple species M. hupehensis has been used as a physiological, and more recently molecular, model to study juvenility173. However, unlike most M. domestica cultivars and other Malus species, M. hupehensis is triploid, and its allopolyploid genome presents significant technical challenges for sequence assembly. The high-quality M. domestica genome sequence will provide a roadmap towards understanding the genetic basis for variance in this trait in M. hupehensis, and ultimately lead to faster and more efficient breeding of new apple cultivars.
Fruit ripening genes and genetics: maturing research
The apple WGS has been a fundamental resource for the determination of key genes involved in the climacteric response and cell wall modification of apple fruit. The attractiveness of apple fruit is due to an array of features (shape, size, color, texture, and aroma), which from the initial phase of fruit development continuously changes through the fruit ripening process174. In climacteric fruit, including apple, fruit ripening is triggered and coordinated by the action of the hormone ethylene. This simple gaseous hydrocarbon is synthesized through the Yang’s cycle that, starting from methionine, produces ethylene through the action of three main enzymes: S-adenosyl-l-methionine synthase (SAMS), 1-aminocyclopropane-1-carboxylic acid synthase (ACS), and 1-aminocyclopropane-1-carboxylic acid oxidase (ACO)175,176. Following synthesis, this hormone is perceived by a series of receptors (ethylene response sensor and ethylene resistant) that initiate a downstream signaling mitogen-activated protein kinase cascade (constitutive triple response—ethylene insensitive 2) activating finally a series of ethylene-dependent responses mediated by ethylene response factor transcription factors177,178. The ethylene competitor 1-methylcyclopropene (1-MCP) has commonly been used to reveal the role of ethylene in controlling the overall ripening process in apple. Investigation of the genome-wide transcriptional signature was enabled by use of two microarrays (iRIPE and WGAA) designed using the information made available by the apple WGS. Through use of these tools, a gene de-repression or de novo activation, was revealed following 1-MCP treatment179. Gene annotation and prediction using the apple WGS enabled detection of possible cross-talk between ethylene and auxin. Specifically, interference at the ethylene receptor level stimulated expression of Aux/IAA and AUXIN RESPONSE FACTOR (ARF) elements involved in the auxin signaling pathway179,180. Upregulation of these genes was further validated through a RNA-seq survey performed to investigate the physiological mechanisms controlling apple superficial scald181. Gene annotation also facilitated the detection of important elements involved in postharvest ripening, as well as in the control of this scald. Furthermore, the apple WGS enabled development of a new microarray (AryANE v1.0)182 that was used to investigate the cell wall modification process during ripening. This research identified a specific pectin methylesterase (MdPME2) as well as elements involved in hemicellulose metabolism183,184.
The apple WGS has also facilitated identification of QTLs associated with control of fruit quality. Genome re-sequencing of 14 apple accessions11 enabled prediction of eSNPs, which were subsequently included in the 20K SNP array. With this SNP array, QTLs associated with mechanical and acoustic components of apple texture were detected via PBA81. The same tool enabled a GWAS that revealed the genetic relationship between texture and the production of volatile organic compounds185. Here, the apple WGS facilitated anchoring of the QTL intervals and cataloging the underlying gene sets.
Further improvement of the quality of the genome assembly and fine-tuning of the gene annotation should enable more precise and informative characterization of the relevant genes involved in the control of important physiological pathways underlying the apple fruit ripening processes, ultimately enabling the development of new cultivars characterized by superior fruit quality.
Fruit color genes and genetics: beyond skin depth with the MYB10 gene
Fruit appearance is a key factor in consumer acceptance186. Red coloration in the skin and flesh of apple fruit has been demonstrated to be controlled genetically (by allelic variation)187,188,189,190, by the environment (differences in light and temperature)191,192 as well as by management (e.g., bagging vs. not bagging fruit). Fruit color is determined by anthocyanin accumulation in the vacuole (mostly cyanidin-3O-galactoside), which is regulated by the MYB1/MYB10 transcription factor in combination with bHLH and a WD40 partners in a protein complex92. MYB1 and MYB10, which are likely to be allelic192, regulate the activity of the genes of the anthocyanin pathway and their expression correlates with anthocyanin accumulation. A locus closely linked to MYB10 has been mapped and associated with red flesh and red skin using biparental populations and GWAS in germplasm collections13,83,189,193,194,195. Interestingly, this red fruit (Rf) locus is orthologous with red color-imparting loci detected in other Rosaceae fruit crops such as cherry, peach and strawberry196. Recently, a robust allele-specific qPCR marker was developed and validated using both apple breeding families and genetically diverse cultivars197. Despite the relative simple genetic control of fruit color (the MYB10 locus has been demonstrated to consistently explain greater than 80% of the phenotypic variance for fruit skin color in populations varying from no red overcolor to much overcolor), the underlying causative variant of this phenotypic variation has not yet been identified. However, the causative variant for red flesh coloration compared to the usual white flesh color has been identified. “Type 1” red flesh has been demonstrated to be due to a repeated motif in the regulatory region upstream of the MYB10 open reading frame190.
The availability of the new GDDH13 reference genome8, together with whole genome re-sequencing data for a range of apple cultivars (old and modern)10,57, opens the prospect of pinpointing the causative variant(s) for red skin color. The GDDH13 WGS assembly offers a unique “haplotype 0” opportunity, as “Golden Delicious” is homozygous for the non-red skin allele. DNA variants located in regulatory motifs upstream of MYB10 are obvious candidates to be examined first, as they might alter MYB10’s activity and consequently influence the downstream activity of the anthocyanin biosynthesis genes. For example, some evidence of epigenetic modification in the promoter region upstream of MYB10 has been demonstrated in striped “Honeycrisp” apples198. More re-sequencing, including using bisulfite-treated DNA, might shed new light on the key regulatory motifs. Such work will need to be complemented with a test of the efficiency of contrasting-effect haplotypes for anthocyanin gene activation, activation of MYB10 itself, and as targets of transcription factors regulating MYB10. One key consideration is that red fruit skin might be associated with more than one allele. In this case, a combination of PBA using a population segregating for red coloration derived from several ancestral sources of red determinism combined with high-throughput genome re-sequencing would be required.
Fruit acidity genes and genetics: epistatic allelic variation in a regulatory pathway
Malate accounts for more than 90% of total fruit organic acids in apple determined by concentration199. Fruit malate content varies widely in Malus accessions: from 0.5 to 22.7 mg/g, with 2.2 times more malate detected in wild species than in M. domestica cultivars200. Enzymes and their coding genes associated with malate biosynthesis and degradation were previously emphasized, until the important role was uncovered of malate transport into the vacuole and its maintenance there201,202,203. Major differences in apple fruit acidity among cultivars were determined to be controlled by a single locus that was mapped to one end of chromosome 16 and designated as the Malic acid (Ma) locus102,204,205. Expression of Ma1 co-segregated with malate content and at least one allele sufficed for increased Ma1 expression, causing a three-fold increase in fruit malate content206.
Exploration of the apple WGS enabled major discoveries after more than a decade of stagnation in characterizing the actual gene underlying apple fruit acidity111,207,208. The Ma locus, explaining 17.0–42.3% of phenotypic variance in fruit pH and titratable acid, was narrowed down to a genomic interval no larger than 150 kb containing 44 genes. Next, the interval was narrowed down further to 65–82 kb containing 12–19 genes. Finally, a G-to-A mutation was identified at 1455 bp of the open reading frame in the Ma1 gene that leads to a premature stop codon, truncating 84 amino acids of an aluminum-activated malate transporter (MdALMTII) protein, and significantly reducing fruit acidity when occurring as a homozygote (mama). However, this natural occurring G-to-A SNP in the coding region of MdALMTII could not fully explain observed variation in fruit acidity among diverse Malus accessions or during fruit developmental changes. This is not surprising, as other acidity QTLs have been reported which occasionally had a size similar to Ma1. A QTL on LG 8 exhibited a similar or higher contribution to fruit acidity in many other segregating families when compared with the LG 16 QTL29,102,209,210, although also a substantial lower contribution has been reported205. This difference in performance might be due to different functional alleles or tightly linked QTLs of different effect segregating among families211. Enabled by whole genome re-sequencing, genetic variation in three genes on chromosome 8, MdSAUR37, MdPP2CH and MdMYB44, were identified and validated. The regulatory pathway of MdSAUR37, MdPP2CH, and MdALMTII was dissected and appears to fully explain the hierarchical epistatic genetic control of apple fruit acidity212.
The expression levels of 3066 genes exhibited significant correlation with malate concentrations during fruit development in “Golden Delicious”213. More than 1300 genes were differentially expressed between cultivars with MaMa/Mama versus mama genotypes214. Several vacuole transporters, V-ATPase, MdVHA-A, MdVHA-B1, MdVHA-E, MdVHP1, MdALMT1, MdALMT6, MdALMT9, and others, some of which under the direct regulation of MYB transcription factors or MdCIPK24-like proteins, were determined to play a pivotal role in determining malate content215,216.
Future research targets utilizing the apple WGS include development of a more detailed network of apple fruit malate metabolism and discovery of further natural genetic variation in the network in diverse Malus germplasm resources. Emphasis should be on the molecular regulation pathways of malate transport to the vacuole, vacuole malate sequestration and depletion during fruit development and post-harvest storage, the effect of different growing environments and functional mutations among cultivars and species. One potential challenge is that in some cultivars acetic acid and tartaric acid can account for 10–31% of total acids210, which together with various classes of aromatics and sugars create the colorful flavors of apples.
Disease resistance genes and genetics: straight to candidate R-genes
The availability of the apple WGS has revolutionized the approaches and reduced the time needed to identify candidate resistance genes. Just over 20 years ago, the mapping of a resistance gene (R gene), which is the first step towards the positional cloning of a gene, was a long and tedious endeavor. Genetic markers such as random amplification of polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP), and restriction fragment length polymorphism (RFLP) were used, generally in combination with bulked segregant analysis (BSA)217, to (roughly) map the R genes. These genetic markers then had to be transformed into SCAR or cleaved amplified polymorphic sequence (CAPS) markers to be useful for marker-assisted selection and/or to start the cloning of an R gene. Around the beginning of the new millennium, genetically mapped SSR markers for apple became available204,218,219,220. In a very short time period, these markers allowed mapping of a series of R genes, such as the apple scab resistance genes Rvi2 and Rvi4221, Rvi5222, Rvi11223, Rvi12224, and Rvi15225, as well as the fire blight resistance genes FB_E and FB_Mf226 and FB_MR5227,228, mildew resistance genes229,230, and several QTLs for resistances (e.g., see ref. 231,232,233).
The establishment of genetic map positions of these resistance genes opened up their potential for cloning and determination of their DNA sequence. Prior to the availability of the first apple WGS of Velasco and co-workers1, “workarounds” were necessary to identify markers closely associated with a gene of interest. For example, during the positional cloning of Rvi15, Galli and co-authors234 developed tightly linked markers using the sequence of a “Florina” bacterial artificial chromosome (BAC) clone of the homolog region. Parravicini and co-authors235 performed an AFLP-BSA approach to clone the fire blight resistance gene FB_E. To our knowledge, Fahrentrapp and co-authors236 were the first to report the use of the apple WGS to fine-map an R gene in apple, FB_MR5. In this case, the apple WGS allowed a rapid saturation with markers of the regions containing the R gene. The time savings can also be recognized by the increase in number of published reports of such research. Before the release of the apple WGS, entire papers were published reporting “only” the fine mapping of an R gene (e.g., see ref. 234,237). In contrast, after the genome release, such work was summarized in relatively short sections of publications, and papers reported not only the fine mapping but also the identification of candidate genes, such as for apple scab (Rvi1, Rvi 5, Rvi 12, and Rvi18)238,239,240,241 and fire blight (FB_MR5, FB_Mfu10228,236,242). The WGS also facilitated research on genetic variation for resistance genes, such as the identification of FB_MR5 analogs in other wild apple species243. Finally, the WGS facilitated determination of the diversity and identity of QTLs from different studies for the same linkage group by clarifying their relative chromosomal position244.
However, identification of many candidate R-gene allele(s) cannot be achieved using the currently available apple WGS, because these resources have not been developed from individuals in the germplasm that carry the resistance alleles. Until now, R-gene alleles have been identified by sequencing BAC clones from libraries of resistance allele-carrying individuals spanning the R-gene regions (e.g., see ref. 234,235,236). The fast progress in sequencing technology and sequence assembly associated with decreasing sequencing cost will soon make the use of BAC libraries obsolete. Instead, whole genome resequencing of individuals carrying an R allele of interest and the subsequent assembly of the specific region carrying that R allele is expected to become the new standard. Furthermore, determining the correct order of repetitive genes will be very important, because resistance genes are often arranged in tandem repeats245.
With the WGS of apple speeding up the process of identifying R genes, translation to crop improvement is also increasing. Genetic markers and knowledge gained from R-gene cloning research is allowing the building of pyramids of R alleles against the same pathogen with different mechanisms of resistance. Such pyramids of R alleles are expected to lead to the development of cultivars with more durable resistances. These alleles could be combined into single individuals by traditional crossing leading to new apple cultivars, or alternatively be introduced into popular cultivars by cisgenic or CRISPR/Cas9 approaches in countries where this is permitted. Protocols in apple for the introduction by CRISPR/Cas9 of large genetic regions (e.g., entire R alleles) are beginning to be reported246,247. However, for some diseases a more straightforward approach might be the targeted knock-out of susceptibility S genes, such as for MdMLO19248 that should lead to development of powdery mildew resistant apple cultivars, at least in the absence of other MLO genes of similar effect249.
Trait-predictive DNA test development: monitoring major genetic factors for breeders
Rapid advances in apple breeding were projected in the publications of both the first and second iterations of the apple WGS1,8. Indeed, progress is being made in the development and application of genomic technologies in the area of trait-predictive tests, although perhaps not as quickly as hoped for by earlier reviewers171,250. The use of SNP arrays10,11,12 and GBS to efficiently screen mapping populations and construct relatively dense genetic maps13,251 has made the mapping of trait loci much less challenging and correspondingly the number of QTLs in the Genome Database for Rosaceae252 has risen rapidly since the availability of the apple WGS. The recent development of a tool utilizing apple resequencing data from breeding germplasm, for the purpose of identifying SNPs that are unique to the accession carrying an attribute of interest, has both increased the efficiency of genetic mapping of trait loci and provided a source of SNPs for use in designing transferable high-throughput markers253 in combination with accurate pedigree records or PBA74,75. Highly reliable diagnostic markers derived from candidate genes for traits can now be readily developed by examining the DNA sequence at MTLs such as major resistance loci and the sequence underlying QTLs254,255.
Early adopters of trait-predictive tests mainly employed simple sequence repeat and SCAR markers256,257,258,259,260 and highly informative SSR markers are still used for tracing pedigrees67,69. Currently, high-throughput SNP-based markers have become the first choice for use in trait-predictive tests for parental and seedling selection by breeders and many programs have moved to develop and introduce the use of such tests80,254,260,261,262,263. As marker-assisted selection (MAS) was introduced into the apple breeding program at the New Zealand Institute for Plant & Food Research Limited (PFR) before the advent of the apple WGS257, PFR researchers across several disciplines were in a sound position to exploit the new knowledge to increase efficiencies and speed up progress towards the PFR goal of multi-resistant new cultivars with pyramided alleles across multiple loci for durable resistance. The PFR scion breeding program employs diagnostic SNP markers to combine alleles at MTLs for resistance to apple scab. Currently, in order to be cost-effective, a phenotypic screen for Rvi2 is followed by MAS for Rvi6. In some families, MAS is applied for additional quality traits, such as red flesh (Myb110a). Both breeding parents and stage 2 selections are routinely screened with a range of markers including those for resistances to scab (Vh8/Rvi8 and Vr/Rvi19), powdery mildew (Pl2), fire blight (Fb-R5), European canker (Rnd1), and woolly apple aphid (Er1, Er2, Er3). The PFR apple rootstock breeding program has employed MAS since 2012, to reduce the population for detailed phenotyping for time-consuming horticultural traits to less than 10% of the initial numbers253,264. Screens applied vary according to year and genetic background of seedlings, but include dwarfing (Dw1, Dw2) resistances to fire blight (FB-R5), European canker (Rnd1), and woolly apple aphid (Er1, Er2, Er3), as well as adventitious rooting265. Nearly all diagnostic markers are applied in the cost-efficient fluorescent probe-based Taqman® format, with more than 25,000 seedlings screened annually in the combined PFR apple breeding programs as a tool to breed better cultivars faster. MAS targeting trait loci for fruit quality and disease resistance is also in routine use in several other apple breeding programs around the world266.
MAS can be and is also routinely employed to reduce numbers of seedlings prior to evaluation by genome-wide selection171. Other types of high-throughput markers have been developed for apple, e.g., KASP™ in the European project FruitBreedomics261 and there are now commercial services for high-throughput marker screening in most areas of the world.
Various avenues of future work are warranted to facilitate the application of trait-predictive tests for MAS. Individual breeding programs will wish to develop markers for specialized traits that are relevant to their own germplasm and breeding goals. New marker options need to be investigated for apple, e.g., GT-Seq, which generates genotypes by NGS from relatively small panels of targeted SNPs267. Further work is required to develop user-friendly bioinformatic tools for access and sharing of sequence data, as well as comprehensive pipelines for the development of specific marker classes, from NGS through to primer/probe sets. Additional development of haplotyping, already initiated with SNP arrays, could be made using re-sequencing and tools such as Beagle268.
Genomic prediction: genome-wide performance forecasting, black box, and beyond
The technology of genomic prediction (GP, also known as genomic selection, genome-wide selection/prediction, and whole genome prediction) was developed in animal genetics to target traits influenced by more than a few large-effect QTLs and soon used widely in animal, forestry and crop improvement269,270,271. Apple was the first horticultural tree crop in which this approach was explored194,272 and implemented269. The premise of GP is that anonymous genome-wide markers are sufficiently dense so that most or all QTLs are in LD with a marker and that most or all QTLs segregate with the use of multi-parent populations. Training populations of multiple families are used to develop prediction models by simultaneously fitting all markers as random effects with a prior distribution273 in contrast to conventional marker-assisted selection which uses only significant large-effect QTLs detected through linkage analysis or GWAS for selection/prediction.
Prediction accuracy, the correlation between true genetic effect and phenotype predicted only from markers in a validation population, is a major driver of GP success but varies greatly (−0.02 to 0.89) across several apple studies194,272,274,275. Consistently, GP accuracy has been demonstrated to be higher than using large-effect QTL-only models for polygenic traits (reviewed by Crossa and co-workers276). Results from apple studies suggest important factors leading to higher accuracy are close genetic relationships between training and validation populations194,274, large training population sizes194,275, small effective size of training and validation populations194,274, high heritability of traits particularly obtained through objective phenotyping194,272, assessment of continuously distributed traits272, and high density of markers272. However, multiple assessments across years of the same experimental unit (i.e., pseudo-replication277) might not efficiently improve heritability and hence not improve accuracy272. The extent of LD in a population is related to effective population size, hence LD and genetic relationship levels are entangled in a population, making it difficult to separate their effects on the accuracy of GP. Genomic relationships among training population individuals can reliably be estimated with a few thousand SNPs278 but many QTLs might not be in strong LD with such a low density of SNPs due to the distribution of SNP and QTL locations across the genome. LD between SNPs and QTLs, and relatedness between training and selection candidates, would decay over generations83. Hence, the accuracy of GP over successive generations would reduce unless GP models are recalibrated, although the equivalent is also needed with any linkage-based prediction method279. To date, population sizes are considerably smaller, and density of genotyping lower, in apple studies compared to those for animals.
The influence of using different prediction models has been explored in apple. Assumption of a Gaussian distribution (i.e., many markers with small effect or approximate quantitative trait model) has been the most common in apple GP studies194,274,280, but other distributions are possible281 and have been used272 although no effect was found on prediction accuracy194,282. Genomic prediction models can combine whole-genome prediction and detection of large-effect QTLs283,284,285 and in apple a Bayesian LASSO model was used to detect regions of high influence for fruit quality traits194. The Gaussian framework, however, allows extension to more complex models276 and has been used to model both additive and non-additive genetic effects, although negligible effect on prediction accuracy of breeding value (additive) or clonal values was reported in apple274 and cherry286. These models were also used to study genotype-by-environment interaction (G×E) for fruit quality traits274,287.
The fundamental aim of GP is greater gain at reduced cost and in less time than conventional strategies276 and considerable increases in gain across animals and crops has been demonstrated271. Use of GP for parental breeding value selection and juvenile clonal value selection was reported in The New Zealand Institute for Plant & Food Research Limited (PFR) apple scion breeding program to bypass the initial stage of juvenile testing by reducing the initial population by 90%269. However, as many traits are of interest in apple288, this strategy might work best when focusing on a single trait that is governed by many loci of minor effect. Selection of parents from the training population provides a strong genetic link with the selection population, to improve prediction accuracy. GP is reportedly in initial use in US apple programs (J. Luby, Uni. Minnesota, pers. comm.).
As GP is a unifying technology across animal and plant breeding289, advances across these systems can provide direction for fundamental discoveries and practical application in apple. The ability to clone apple individuals provides opportunities to counteract low prediction accuracy due to small sample sizes or large experimental variation. Improvement in accuracy from multi-environment and multi-trait prediction models found in other crops276,290,291 needs to be verified for apple. Inclusion of biological information, such as previously identified large-effect QTLs and/or extension to crop growth (development) or functional structural models might also help increase prediction accuracy271,275,292,293. The biallelic nature of SNPs suggests combining linked SNPs into haplotype blocks (haploblocks) is likely to be more informative of the variability at a QTL and could decrease hidden variability, hence leading to higher prediction accuracy271,294,295. While the use of sequence information to build GP models suggests higher accuracy because the causal variation should be directly included in the model, the value of this extension in apple needs to be verified as populations are highly structured69 such that large haploblocks could be efficiently tagged with a lower density of markers271,296. Fast-track breeding via transgenesis297,298 or induced through a plant virus vector299 might enable full realization of GP potential in apple. While SSRs were used to select for a favorable QTL allele and against unfavorable genetic background over five generations in seven years300, GP can be used to identify elite progeny across a range of traits. Implementation of GP for apple improvement might require modification of breeding programs and stochastic simulation can be used to evaluate benefits of different strategies290.
The ability of GP to connect otherwise unconnected populations without the need for clonal replication287,301 provides the foundation for combining data from small local breeding programs to improve accuracy, dissect G×E, predict genetic potential across a global environment, and use as a reference for other research. This approach can therefore leverage the latent value of historically collected trial data. The RosBREED project assembled a large set of apple phenotypic and genotypic data from across 14 global locations (three USA, seven EU, two New Zealand, and two Australian) to develop a global GP model for fruit sweetness and acidity and evaluate the accuracy of predictions. Bioinformatics tools are being developed to manage and deliver the technology so that the performance of new germplasm can then be routinely predicted.
Epigenetics: scratching the surface
Access to a near-complete apple WGS provides the foundation for epigenetic studies. A key area unlocked by the availability of the high-quality GDDH13 WGS is the study of sports (i.e., clones showing a novel phenotype) and, associated with this, the study of epigenetic mechanisms and their contribution to important traits such as fruit size and color. By definition, epigenetic changes result in heritable changes in gene expression that cannot be explained by changes in the DNA sequence. To be able to study true epigenetic events, one has to ensure that there are as few genetic changes as possible in the genome under examination. Therefore, apple sports are therefore an excellent starting point to study epigenetics in plants. Although some sports might be the result of genetic changes (such as mutations induced by transposable elements302, others might vary only at the epigenetic level and more specifically at the DNA methylation level.
The main reason for generating the apple reference genome GDDH13 was to enable an understanding of the epigenetic mechanisms involved in regulating fruit size. This approach was successful, as the research generated a list of genes likely involved in fruit size regulation8. Currently, there is no direct impact of apple epigenetics research on crop improvement. Future studies using epigenetic markers on sports and on larger populations will enable assessment of the contribution of DNA methylation to traits of interest. Considering that DNA methylation also influences fruit color intensity and patterning in apple sports198,303, it is likely that we have only started to scratch the surface of the contribution of epigenetics to traits of economic importance.
Several epigenetic studies are expected to profit from the availability of high-quality apple WGSs in the near future. The study of the genetic and epigenetic mechanisms influencing phenotypic changes of apple sports is straightforward with a good genome assembly and will allow the rapid identification of genes involved in important agronomic traits, including fruit color and shape, disease resistance and tree architecture. Chemical-induced or targeted changes in DNA methylation patterns could be used to produce novel traits of interest by gene demethylation. DNA methylation changes can also be used to induce the artificial mobilization of transposable elements304, which results in a powerful tool for gene discovery (via tagged mutations) and the generation of phenotypic diversity.
Genome database for Rosaceae: the researcher’s toolbox
Apple researchers exploiting the apple WGS are well provisioned with data and tools housed in the Genome Database for Rosaceae (GDR). The GDR305 (https://www.rosaceae.org) is the publicly accessible, central repository and data-mining resource for genomics, genetics, and breeding data of the Rosaceae family, which includes apple as well as other economically important crops. The GDR is built using Tripal306,307, a resource-efficient, open-source platform for online biological database construction. Prior to the availability of the first WGS in 2012, the GDR contained mostly EST data, genetic markers, and genetic maps. The new data types currently included in the GDR include multiple WGSs, reference transcriptomes, QTLs, SNP array data, and phenotypic and genotypic data. Increased data from the worldwide apple research community, combined with active curation, analyses, and tool development, has resulted in significant expansion of the GDR during the last few years. Described below are the currently available data and interfaces, with a focus on new features.
The physical structure of the apple WGS, as described by its nucleotide sequence, is the foundation of apple genomic resources in the GDR. Genomic data include the GDDH13 WGS8 along with three historical versions of the heterozygous apple genome assembly1. Although no chromosomal sequence is available for these heterozygous apple genome assemblies, the sequences of overlapping contigs are available, which in turn have been aligned to the chromosome using markers from the genetic map. For the Golden Delicious v1.0 WGS, however, a set of four pseudo-haplotype assemblies (primary and alternatives 1, 2, and 3) with chromosomal sequences, derived from the contigs of the original v1.0 assembly, is available for download. The primary pseudo-haplotype data is also available in the Search Genes page and JBrowse308 in the GDR. Researchers can readily examine in fine detail how the apple genome compares physically with those of other crops. The most recent apple WGS is being used in synteny analysis with eight other Rosaceae whole genome assemblies using MCScanX309, with the results available through the new Synteny Viewer.
Another strong focus of the GDR is apple gene function. Since 2017, the community database has provided an apple reference transcriptome (M. domestica GDR RefTrans V1) that combines published RNA-Seq and EST data that are computationally annotated for homology to genes of other plant species and assignment of InterPro domains310 and GO terms311,312. The data can be accessed in various ways: through the Species page, the Search Genes and Transcripts page, the JBrowse308 tool, or BLASTX313 tool.
Genetic linkage, marker and trait locus information needs are well served. The GDR contains 108 genetic maps for Malus, which can be viewed and compared through a new graphic interface, MapViewer. Currently, the GDR contains 2.6 million genetic Malus markers, of which 99.7% are SNPs while more than 7000 are other marker types. SNP data include those of the 9K10, 20K11, and 480K12 apple arrays. Most of the remaining SNPs are those identified in silico from accession resequencing conducted during development of those SNP arrays. The SNP data are available as JBrowse tracks, downloadable files, and from the Marker Search page. The Marker Search page has a new Filter-by-Trait name feature, which enables researchers to find markers associated with QTLs. In addition, the SSR and SNP genotype search pages contain data from ten genotyping projects. Trait locus data in the GDR includes 1528 QTLs and 36 MTLs for 143 horticultural traits of apple. Although impressive, some caution is necessary in considering these numbers. They are based on the accumulation of QTLs as independently reported in the literature and therefore might include redundancies across studies or treatments (including years) within a single study. In addition, some caution is be needed in using reported QTL information. Their assignment might arise from different levels of statistical significance; regrettably, it is often not specified whether a genome-wide or a chromosome-wide threshold was applied. Some reported QTLs were from an interval mapping procedure without subsequent co-factor analysis. Such QTLs have increased risk of being spurious or being assigned to an incorrect position, while other true QTLs might remain hidden314,315.
Phenotypic data for breeding germplasm are also accessible on the GDR, from publicly funded projects such as RosBREED78. In addition to the Search Trait Evaluation page, the public breeding data can be accessed using the Breeding Information Management System (BIMS). BIMS allows breeders to store, manage, archive, and analyze their private or public breeding data.
The data of the original apple WGS1 enabled the GDR to integrate various data types. Functional annotation of gene models using sequence similarity and synteny analyses provided a first step in data integration across species. Alignment of transcripts and markers, used in genotyping and QTL mapping, to the WGS, enables researchers to utilize data of different types originating from various species and disciplines. Future targets include integrating new types of data such as pan-genome data, epigenome data, and phenomics data, as well as data standardization and further tool development. In this way, the GDR will continue to enable researchers to open new frontiers in understanding the structure and function of the apple genome and its components.
Conclusion
Over the past decade, apple WGSs have had an enormous impact on our understanding of apple biological functioning, trait physiology and inheritance, leading to practical applications for improving this highly valued crop. The first apple WGS reported in 20101, v1.0, was valuable as the initial reference for sequence information, fine mapping, gene discovery, variant discovery, and tool development. As the research community exploited this resource, the need for improvements in the genome assembly became increasingly apparent. The new GDDH13 assembly reported in 20178 provided many of those improvements, and research leaps ensued. In contrast to a decade ago, causal gene identities for phenotypes of fundamental or practical interest can today be discovered rapidly. Genome-wide polymorphisms are readily screened for hundreds to thousands of germplasm individuals. Genetic maps are constructed readily and QTLs are quickly related to candidate genes and/or converted into diagnostic tests for breeding use. We now understand the species, geographical, and genomic origins of domesticated apple more precisely, as well as the crop’s relationship to potentially valuable sources of alleles in wild relatives. Availability of the WGS is not only turbo-charging the conduct of these classical research steps to crop improvement, but is also driving innovative methods of achieving more durable, environmentally sound, productive, and consumer-desirable apple crop production.
Research collaboration on apple genomics and genetics, already vibrant across an international community, has much synergistic value. The physical map of apple’s 17 chromosomes is a shared foundation that unites diverse fields of apple science. Although it can be viewed in many ways, each chromosome has the same physical entity for any researcher. The apple WGS with its set number of chromosomes and their lengths delineates the boundaries of what has arisen through natural means to coalesce as the “blueprint” of an apple tree. Researchers worldwide are resolutely identifying the roles and influences, intra-genomic interactions, and germplasm distributions of variants of chromosomal modules—the genes, motifs, trait loci, haploblocks, base pairs and so on—all using the WGS of apple. Integration of the apple transcriptome, proteome, metabolome, epigenome, QTLome316, and so on, across experiments and experimental material will surely expand our scientific understanding of apple. The sheer amount of multi-dimensional genomic and downstream data generated has elevated the need and opportunities for their coordination to a new level. To sustain such collaborative advances, the GDR now serves as the research hub for the international apple genomics community. We should also think beyond apple: exploiting the genomic synteny of apple with crop relatives in the Rosaceae family has only just begun, and holds much potential for wider collaboration and research breakthroughs.
Advances are needed in upgrades to the base genome sequence data pool. The reference WGS should be improved to ensure its completeness and accuracy. A first step could be noting in genome browsers any suspicious regions identified by comparisons between the WGS and high-quality genetic linkage maps. Given the possibility that the doubled-haploid individual underlying the current reference WGS is missing parts of its genome compared to typical heterozygous cultivars8, there is still a need for a reference WGS representing the complete cultivated apple genome. In any case, the linear or “vertical” representation of the apple WGS needs to be extended laterally to capture sequence variation along each chromosome. Development of the apple pan-genome would be a clear way forward for describing the current allelic variation of cultivated apple. Expansion of the pan-genome to encompass the Malus genus is a further compelling target as M. domestica is able to be crossed with dozens of other species.
Advances are also needed in use of WGS-related data to answer fundamental and applied research questions. There is an urgent need for an apple gene atlas describing the roles and relationships of apple’s 42,140 genes, as currently more than half the putative protein-coding genes of apple have unknown functions. Another challenge is understanding the interactions of genes with the external world, be they via transient methylation, inherited epigenetic changes, or statistically trackable and exploitable interactions of G×E (×M, crop management, and ×A, age or ontogeny of plants). Genomic prediction approaches promise a foundation for genotype and environment matching. A major leap in analytical capabilities is required to gain information on the vast reservoir of alleles that already contribute to the phenotypes of cultivated and wild apple trees. For apple WGS information to be able to directly support new cultivar development, user-friendly bioinformatics solutions are needed. Tools are required for ready conversion of trait-predictive DNA tests to commonly used and new genotyping platforms and for whole-genome-profiling diagnostics. Streamlined data curation workflows would enable any given breeding program to benefit, by translating genotypic data into genetic information that supports breeding decisions. Intuitive analytical tools are needed to track, over generations, all alleles of interest linked along each chromosome. Ultimately, such tools will facilitate understanding of genetic relationships across germplasm and empower “Breeding by Design”317 for breeders to access allelic diversity in available germplasm and design and target whole-genome ideotypes built from alleles of interest.
The curious apple scientist thirsts for knowledge—for every question answered, many more are raised. The apple WGSs have already enabled the answering of numerous questions. Tangible impacts have resulted because of the integration of apple genomics and genetics research with breeding. We hope the examples of apple WGS impacts inspire creative new ideas that spur further advances in understanding and improving apple.
References
Velasco, R. et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 42, 833–839 (2010).
Bolger, M. E. et al. Plant genome sequencing—applications for crop improvement. Curr. Opin. Biotechnol. 26, 31–37 (2014).
Root W. H. & Barret D. M. in Processing Fruits, Science and Technology 2nd edn (eds Barret D. M., Somogyi L. & Hosahalli R.) (CRC Press, Boca Raton, FL, 2004).
Noiton, D. A. M. & Alspach, P. A. Founding clones, inbreeding, coancestry, and status number of modern apple cultivars. J. Am. Soc. Hortic. Sci. 121, 773–782 (1996).
Evans, K. M. et al. Genotyping of pedigreed apple breeding material with a genome-covering set of SSRs: trueness-to-type of cultivars and their parentages. Mol. Breed. 28, 535–547 (2011).
Evans, K. M., Barritt, B. H., Konishi, B. S., Brutcher, L. J. & Ross, C. F. ‘WA 38’ Apple. HortScience 47, 1177–1179 (2012).
Howard, N. P. et al. Elucidation of the ‘Honeycrisp’ pedigree through haplotype analysis with a multi-family integrated SNP linkage map and a large apple (Malus×domestica) pedigree-connected SNP data set. Hortic. Res. 4, 17003 (2017).
Daccord, N. et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 49, 1099–1106 (2017).
Li, X. et al. Improved hybrid de novo genome assembly of domesticated apple (Malus × domestica). Gigascience 5, 35 (2016).
Chagné, D. et al. Genome-wide SNP detection, validation, and development of an 8K SNP array for apple. PLoS ONE 7, e31745 (2012).
Bianco, L. et al. Development and validation of a 20K single nucleotide polymorphism (SNP) whole genome genotyping array for apple (Malus × domestica Borkh). PLoS ONE 9, e110377 (2014).
Bianco, L. et al. Development and validation of the Axiom(®) Apple 480K SNP genotyping array. Plant J. 86, 62–74 (2016).
Gardner, K. M. et al. Fast and cost-effective genetic mapping in apple using next-generation sequencing. G3 4, 1681–1687 (2014).
Migicovsky, Z. et al. Genome to phenome mapping in apple using historical data. Plant Genome 9, 2 (2016).
Migicovsky, Z., Li, M., Chitwood, D. H. & Myles, S. Morphometrics reveals complex and heritable apple leaf shapes. Front. Plant Sci. 8, 2185 (2018).
Amyotte, B., Bowen, A. J., Banks, T., Rajcan, I. & Somers, D. J. Mapping the sensory perception of apple using descriptive sensory evaluation in a genome wide association study. PLoS ONE 12, e0171710 (2017).
McClure, K. A. et al. A genome-wide association study of apple quality and scab resistance. Plant Genome 11, 1 (2018).
Money, D., Migicovsky, Z., Gardner, K. & Myles, S. LinkImputeR: user-guided genotype calling and imputation for non-model organisms. BMC Genom. 18, 523 (2017).
Wickland, D. P., Battu, G., Hudson, K. A., Diers, B. W. & Hudson, M. E. A comparison of genotyping-by-sequencing analysis methods on low-coverage crop datasets shows advantages of a new workflow, GB-eaSy. BMC Bioinform. 18, 586 (2017).
Motazedi E., Finkers R., Maliepaard C. & Ridder D. Family-based haplotype estimation and allele dosage correction for polyploids using short sequence reads. bioRxiv. https://doi.org/10.1101/318196 (2018).
Qi, P. et al. UGbS-Flex, a novel bioinformatics pipeline for imputation-free SNP discovery in polyploids without a reference genome: finger millet as a case study. BMC Plant Biol. 18, 117 (2018).
N’Diaye, A. et al. Single marker and gaplotype-based association analysis of semolina and pasta colour in elite durum wheat breeding lines using a high-density consensus map. PLoS ONE 12, e0170941 (2017).
Bourke, P. M. et al. Multi-environment QTL analysis of plant and flower morphological traits in tetraploid rose. Theor. Appl. Genet. 131, 2055–2069 (2018).
Voorrips, et al. PediHaplotyper: software for consistent assignment of marker haplotypes in pedigrees. Mol. Breed. 36, 119 (2016).
Verde, I. et al. The Peach v2.0 release: high-resolution linkage mapping and deep resequencing improve chromosome-scale assembly and contiguity. BMC Genom. 18, 225 (2017).
Vanderzande, S. et al. High-quality, genome-wide SNP genotypic data for pedigreed germplasm of the diploid outbreeding species apple, peach, and sweet cherry through a common workflow. bioRxiv. https://doi.org/10.1101/514281 (2019).
Antanaviciute, L. et al. Development of a dense SNP-based linkage map of an apple rootstock progeny using the Malus Infinium whole genome genotyping array. BMC Genom. 13, 203 (2012).
Troggio, M. et al. Evaluation of SNP data from the Malus Infinium array identifies challenges for genetic analysis of complex genomes of polyploid origin. PLoS ONE 8, e67407 (2013).
Ma, B. et al. Construction of a high density linkage map and its application in the identification of QTLs for soluble sugar and organic acid components in apple. Tree Genet. Genomes 12, 1 (2016).
Di Pierro, E. A. et al. A high-density, multi-parental SNP genetic map on apple validates a new mapping approach for outcrossing species. Hortic. Res. 3, 16057 (2016).
Jia, H.-M. et al. The red bayberry genome and genetic basis of sex determination. Plant. Biotechnol. J. 17, 397–409 (2018).
Gao, Z. S. et al. Genomic cloning and linkage mapping of the Mal d 1 (PR-10) gene family in apple (Malus domestica). Theor. Appl. Genet. 111, 171–183 (2005).
Zheng, L. et al. Genome-wide identification and expression analysis of brassinosteroid biosynthesis and metabolism genes regulating apple tree shoot and lateral root growth. J. Plant. Physiol. 231, 68–85 (2018).
Dong, Q. et al. Genome-wide analysis and cloning of the apple stress-associated protein gene family reveals MdSAP15, which confers tolerance to drought and osmotic stresses in transgenic Arabidopsis. Int. J. Mol. Sci. https://doi.org/10.3390/ijms19092478 (2018).
Cornille, A., Giraud, T., Smulders, M. J. M., Roldán-Ruiz, I. & Gladieux, P. The domestication and evolutionary ecology of apples. Trends Genet. 30, 57–65 (2014).
Cornille, A. et al. New insight into the history of domesticated apple: secondary contribution of the European wild apple to the genome of cultivated varieties. PLoS Genet. 8, e1002703 (2012).
Coart, E., Glabeke, S. V., Loose, M. D., Larsen, A. S. & Roldán‐Ruiz, I. Chloroplast diversity in the genus Malus: new insights into the relationship between the European wild apple (Malus sylvestris (L.) Mill.) and the domesticated apple (Malus domestica Borkh.). Mol. Ecol. 15, 2171–2182 (2006).
Cornille, A. et al. Anthropogenic and natural drivers of gene flow in a temperate wild fruit tree: a basis for conservation and breeding programs in apples. Evol. Appl. 8, 373–384 (2015).
Feurtey, A., Cornille, A., Shykoff, J. A., Snirc, A. & Giraud, T. Crop-to-wild gene flow and its fitness consequences for a wild fruit tree: Towards a comprehensive conservation strategy of the wild apple in Europe. Evol. Appl. 10, 180–188 (2017).
Brandenburg, J.-T. et al. Independent introductions and admixtures have contributed to adaptation of European maize and its American counterparts. PLoS Genet. 13, 1–30 (2017).
Burgarella, C. et al. Adaptive introgression: an untapped evolutionary mechanism for crop adaptation. Front. Plant Sci. 10, 4 (2019).
Micheletti, D. et al. On the evolutionary history of the domesticated apple. Nat. Genet. 43, 1044–1045 (2011).
Pagliarani, G. et al. Genomic organisation of the Mal d 1 gene cluster on linkage group 16 in apple. Mol. Breed. 29, 759–778 (2012).
Verde, I. et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 45, 487–494 (2013).
Chagné, D. et al. The draft genome sequence of European pear (Pyrus communis L. ‘Bartlett’). PLoS ONE 9, e92644 (2014).
Aravanopoulos F. A., Ganopoulos I. & Tsaftaris A. Population and conservation genomics in forest and fruit trees. Adv. Bot. Res. 74, 125–155 (2015).
Michael, T. P. & VanBuren, R. Progress, challenges and the future of crop genomes. Curr. Opin. Plant Biol. 24, 71–81 (2015).
Hazzouri, K. M. et al. Whole genome re-sequencing of date palms yields insights into diversification of a fruit tree crop. Nat. Commun. 6, 8824 (2015).
Huang, J. et al. The jujube genome provides insights into genome evolution and the domestication of sweetness/acidity taste in fruit trees. PLoS Genet. 12, 1–20 (2016).
Cao, K. et al. Genome-wide association study of 12 agronomic traits in peach. Nat. Commun. 7, 13246 (2016).
Besnard, G., Terral, J.-F. & Cornille, A. On the origins and domestication of the olive: a review and perspectives. Ann. Bot. 121, 385–403 (2017).
Teh, B. T. et al. The draft genome of tropical fruit durian (Durio zibethinus). Nat. Genet. 49, 1633 (2017).
Flowers, J. M. et al. Cross-species hybridization and the origin of North African date palms. Proc. Natl Acad. Sci. 116, 1651–1658 (2019).
Yu, Y. et al. Genome re-sequencing reveals the evolutionary history of peach fruit edibility. Nat. Commun. 9, 5404 (2018).
Gaut, B. S., Díez, C. M. & Morrell, P. L. Genomics and the contrasting dynamics of annual and perennial domestication. Trends Genet. 31, 709–719 (2015).
Neale, D. B., Martínez-García, P. J., De La Torre, A. R., Montanari, S. & Wei, X.-X. Novel insights into tree biology and genome evolution as revealed through genomics. Annu. Rev. Plant. Biol. 68, 457–483 (2017).
Duan, N. et al. Genome re-sequencing reveals the history of apple and supports a two-stage model for fruit enlargement. Nat. Commun. 8, 249 (2017).
Yao, J.-L. et al. A microRNA allele that emerged prior to apple domestication may underlie fruit size evolution. Plant J. 84, 417–427 (2015).
Ballester, A.-R. et al. Transcriptomic response of resistant (PI613981–Malus sieversii) and susceptible (“Royal Gala”) genotypes of apple to blue mold (Penicillium expansum) infection. Front. Plant Sci. 8, 1981 (2017).
Norelli, J. L. et al. Genotyping-by-sequencing markers facilitate the identification of quantitative trait loci controlling resistance to Penicillium expansum in Malus sieversii. PLoS ONE 12, e0172949 (2017).
Meyer, R. S., DuVal, A. E. & Jensen, H. R. Patterns and processes in crop domestication: an historical review and quantitative analysis of 203 global food crops. New Phytol. 196, 29–48 (2012).
Roux, C. et al. Shedding light on the grey zone of speciation along a continuum of genomic divergence. PLoS Biol. 14, e2000234 (2016).
Racimo, F., Marnetto, D. & Huerta-Sánchez, E. Signatures of archaic adaptive introgression in present-day human populations. Mol. Biol. Evol. 34, 296–317 (2017).
Medina, P., Thornlow, B., Nielsen, R. & Corbett-Detig, R. Estimating the timing of multiple admixture pulses during local ancestry inference. Genetics 210, 1089–1107 (2018).
Skov, L. et al. Detecting archaic introgression using an unadmixed outgroup. PLoS Genet. 14, 1–15 (2018).
Way, R. D. et al. Apples (Malus). Acta Hortic 290, 3–46 (1991).
Urrestarazu, J. et al. Analysis of the genetic diversity and structure across a wide range of germplasm reveals prominent gene flow in apple at the European level. BMC Plant Biol. 16, 130 (2016).
Lassois, L. et al. Genetic diversity, population structure, parentage analysis, and construction of core collections in the French apple germplasm based on SSR markers. Plant Mol. Biol. Rep. 34, 827–844 (2016).
Larsen, B. et al. Population structure, relatedness and ploidy levels in an apple gene bank revealed through genotyping-by-sequencing. PLoS ONE 13, e0201889 (2018).
Muranty, H. et al. Reconstruction of multi-generation pedigrees involving numerous old apple cultivars thanks to whole-genome SNP data. 9th International Rosaceae Genomics Conference (Nanjing, 2018).
Howard, N. P. et al. Collaborative project to identify direct and distant pedigree relationships in apple. 9th International Rosaceae Genomics Conference (Nanjing, 2018).
Peace, C., Piaskowski, J. & Vanderzande, S. Visualizing the genetics of elite genomes. 9th International Rosaceae Genomics Conference (Nanjing, 2018).
Verma, S. & Whitaker, V. M. Prediction of QTL genotypes and trait phenotypes using FlexQTLTM: a pedigree-based analysis approach. J. Plant Biol. Crop Res. 2, 1006 (2018).
van de Weg, W. E. et al. Pedigree genotyping: a new pedigree-based approach of QTL identification and allele mining by exploiting breeding material. Acta Hortic. 708, 483–488 (2006).
Bink, M. Ca. M. et al. Bayesian QTL analyses using pedigreed families of an outcrossing species, with application to fruit firmness in apple. Theor. Appl. Genet. 127, 1073–1090 (2014).
Peace, C. P., Luby, J. J., van de Weg, W. E., Bink, M. C. A. M. & Iezzoni, A. F. A strategy for developing representative germplasm sets for systematic QTL validation, demonstrated for apple, peach, and sweet cherry. Tree Genet. Genomes 10, 1679–1694 (2014).
Iezzoni, A. et al. RosBREED: Enabling marker-assisted breeding in Rosaceae. Acta Hortic. 859, 389–394 (2010).
Iezzoni, A. et al. RosBREED2: progress and future plans to enable DNA-informed breeding in the Rosaceae. Acta Hortic. 1172, 115–118 (2017).
Laurens, F. et al. Review of fruit genetics and breeding programmes and a new European initiative to increase fruit breeding efficiency. Acta Hortic. 929, 95–102 (2012).
Laurens, F. et al. An integrated approach for increasing breeding efficiency in apple and peach in Europe. Hortic. Res. 5, 11 (2018).
Di Guardo, M. et al. Deciphering the genetic control of fruit texture in apple by multiple family-based analysis and genome-wide association. J. Exp. Bot. 68, 1451–1466 (2017).
Myles, S. et al. Association mapping: critical considerations shift from genotyping to experimental design. Plant Cell 21, 2194–2202 (2009).
Kumar, S. et al. Novel genomic approaches unravel genetic architecture of complex traits in apple. BMC Genom. 14, 393 (2013).
Urrestarazu, J. et al. Genome-wide association mapping of flowering and ripening periods in apple. Front. Plant Sci. 8, 1923 (2017).
Newcomb, R. D. et al. Analyses of expressed sequence tags from apple. Plant Physiol. 141, 147–166 (2006).
Gasic, K. et al. Characteristics and transferability of new apple EST-derived SSRs to other Rosaceae species. Mol. Breed. 23, 397–411 (2009).
Zhang, J. et al. An optimized TRV-based virus-induced gene silencing protocol for Malus crabapple. Plant Cell Tissue Organ Culture 126, 499–509 (2016).
Schaffer, R. J. et al. A genomics approach reveals that aroma production in apple is controlled by ethylene predominantly at the final step in each biosynthetic pathway. Plant Physiol. 144, 1899–1912 (2007).
Atkinson, R. G. et al. Downregulation of POLYGALACTURONASE1 alters firmness, tensile strength and water loss in apple (Malus × domestica) fruit. BMC Plant Biol. 12, 129 (2012).
Ireland, H. S. et al. Apple SEPALLATA1/2-like genes control fruit flesh development and ripening. Plant J. Cell Mol. Biol. 73, 1044–1056 (2013).
James, D. J., Passey, A. J., Barbara, D. J. & Bevan, M. Genetic transformation of apple (Malus pumila Mill.) using a disarmed Ti-binary vector. Plant Cell Rep. 7, 658–661 (1989).
Espley, R. V. et al. Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J. 49, 414–427 (2007).
Yue, C. & Tong, C. Consumer preferences and willingness to pay for existing and new apple varieties: evidence from apple tasting choice experiments. Horttechnology. 21, 376–383 (2011).
Seppä, L., Latvala, T., Akaichi, F., Gil, J. M. & Tuorila, H. What are domestic apples worth? Hedonic responses and sensory information as drivers of willingness to pay. Food Qual. Prefer. 43, 97–105 (2015).
Waltz, E. Nonbrowning GM apple cleared for market. Nat. Biotechnol. 33, 326–327 (2015).
Ruffell, D. Court of Justice extends the GMO Directive to gene-edited organisms. FEBS Lett. 592, 3653–3657 (2018).
Jouanin, A., Boyd, L., Visser, R. G. F. & Smulders, M. J. M. Development of wheat with hypoimmunogenic gluten obstructed by the gene editing policy in Europe. Front. Plant Sci. 9, 1523 (2018).
Lespinasse, Y. & Le pommier. in Amélioration des espèces végétales cultivées, objectifs et critères de sélection (eds Gallais, A. & Bannerot, H.) 579–594 (INRA Editions, Paris, 1992).
Watkins, R. & Spangelo, L. P. Components of genetic variance for plant survival and vigor of apple trees. Theor. Appl. Genet. 40, 195–203 (1970).
Oraguzie, N. C., Hofstee, M. E., Brewer, L. R. & Howard, C. Estimation of genetic parameters in a recurrent selection program in apple. Euphytica 118, 29–37 (2001).
Lawson, D. M., Hemmat, M. & Weeden, N. F. The use of molecular markers to analyze the inheritance of morphological and developmental traits in apple. J. Am. Soc. Hortic. Sci. 120, 532–537 (1995).
Liebhard, R., Kellerhals, M., Pfammatter, W., Jertmini, M. & Gessler, C. Mapping quantitative physiological traits in apple (Malus × domestica Borkh.). Plant Mol. Biol. 52, 511–526 (2003).
Segura, V., Denancé, C., Durel, C.-E. & Costes, E. Wide range QTL analysis for complex architectural traits in a 1-year-old apple progeny. Genome 50, 159–171 (2007).
Segura, V., Durel, C.-E. & Costes, E. Dissecting apple tree architecture into genetic, ontogenetic and environmental effects: QTL mapping. Tree Genet. Genomes 5, 165–179 (2009).
Foster, T. M. & Aranzana, M. J. Attention sports fans! The far-reaching contributions of bud sport mutants to horticulture and plant biology. Hortic. Res. 5, 44 (2018).
Lapins, K. Inheritance of compact growth type in apple. J. Am. Soc. Hortic. Sci. 101, 133–135 (1976).
Conner, P. J., Brown, S. K. & Weeden, N. F. Randomly amplified polymorphic DNA-based genetic linkage maps of three apple cultivars. J. Am. Soc. Hortic. Sci. 122, 350–359 (1997).
Conner, P. J., Brown, S. K. & Weeden, N. F. Molecular-marker analysis of quantitative traits for growth and development in juvenile apple trees. Theor. Appl. Genet. 96, 1027–1035 (1998).
Kenis, K. & Keulemans, J. QTL analysis of growth characteristics in apple. Acta Hortic. 663, 369–374 (2004).
Moriya, S., Okada, K., Haji, T., Yamamoto, T. & Abe, K. Fine mapping of Co, a gene controlling columnar growth habit located on apple (Malus × domestica Borkh.) linkage group 10. Plant Breed. 131, 641–647 (2012).
Bai, Y. et al. A natural mutation-led truncation in one of the two aluminum-activated malate transporter-like genes at the Ma locus is associated with low fruit acidity in apple. Mol. Genet. Genomics. 287, 663–678 (2012).
Baldi, P. et al. Genetic and physical characterisation of the locus controlling columnar habit in apple (Malus × domestica Borkh.). Mol. Breed. 31, 429–440 (2013).
Wolters, P. J., Schouten, H. J., Velasco, R., Si-Ammour, A. & Baldi, P. Evidence for regulation of columnar habit in apple by a putative 2OG-Fe(II) oxygenase. New Phytol. 200, 993–999 (2013).
Otto, D., Petersen, R., Brauksiepe, B., Braun, P. & Schmidt, E. R. The columnar mutation (“Co gene”) of apple (Malus × domestica) is associated with an integration of a Gypsy-like retrotransposon. Mol. Breed. 33, 863–880 (2014).
Wolters, P. Corrigendum: evidence for regulation of columnar habit in apple by a putative 2OG-Fe (II) oxygenase (vol 200, pg 993, 2013). New Phytol. 207, 928–928 (2015).
Okada, K. et al. Expression of a putative dioxygenase gene adjacent to an insertion mutation is involved in the short internodes of columnar apples (Malus × domestica). J. Plant. Res. 129, 1109–1126 (2016).
Krost, C., Petersen, R. & Schmidt, E. R. The transcriptomes of columnar and standard type apple trees (Malus × domestica)—a comparative study. Gene 498, 223–230 (2012).
Foster, T. M., McAtee, P. A., Waite, C. N., Boldingh, H. L. & McGhie, T. K. Apple dwarfing rootstocks exhibit an imbalance in carbohydrate allocation and reduced cell growth and metabolism. Hortic. Res. 4, 17009 (2017).
Wang, L. et al. The isolation of the IGT family genes in Malus × domestica and their expressions in four idiotype apple cultivars. Tree Genet. Genomes 14, 46 (2018).
Hollender, C. A. & Dardick, C. Molecular basis of angiosperm tree architecture. New Phytol. 206, 541–556 (2015).
Pallas B., et al. Multi-scale high-throughput phenotyping of architectural and functional traits in field/orchard conditions: application to the genotypic variability in an apple tree core-collection under contrasted watering regimes. Hortic. Res. in press (2019).
Glenn, D. M. An analysis of ash and isotopic carbon discrimination (Δ13C) methods to evaluate water use efficiency in apple. Sci. Hortic. 171, 32–36 (2014).
Zhang, L. et al. Role of abscisic acid (aba) in modulating the responses of two apple rootstocks to drought stress. Pak. J. Bot. 46, 117–134 (2014).
Tworkoski, T. & Fazio, G. Hormone and growth interactions of scions and size-controlling rootstocks of young apple trees. Plant Growth Regul. 78, 105–119 (2016).
Bassett, C. L., Glenn, D. M., Forsline, P. L., Wisniewski, M. E. & Farrell, R. E. Characterizing water use efficiency and water deficit responses in apple (Malus ×domestica Borkh. and Malus sieversii Ledeb.) M. Roem. HortScience 46, 1079–1084 (2011).
Kim, M. S. & Ko, K. C. Relation of bitter pit development with mineral nutrients, cultivars, and rootstocks in apples (Malus domestica Borkh). Korean J. Hortic. Sci. Technol. 22, 43–49 (2014).
Koutinas, N., Roubos, K., Sotiropoulos, T. & Therios, I. Response of the apple rootstocks ‘M 9’, ‘M 26’ and ‘MM 106’ to boron toxicity. Acta Hortic. 2013; 471–474.
Fallahi, E. & Mohan, S. K. Influence of nitrogen and rootstock on tree growth, precocity, fruit quality, leaf mineral nutrients, and fire blight in ‘Scarlet Gala’ apple. Horttechnology. 10, 589–592 (2000).
Chun, I. J. & Andong, N. U. Influence of nutrient level and rootstock on fruit quality of ‘Fuji’ apple trees. J. Korean Soc. Hortic. Sci. 45, 252–255 (2004).
Neilsen, G. & Hampson, C. ‘Honeycrisp’ apple leaf and fruit nutrient concentration is affected by rootstock during establishment. J. Am. Pomol. Soc. 68, 178–189 (2014).
Reig, G. et al. Horticultural performance and elemental nutrient concentrations on ‘Fuji’ grafted on apple rootstocks under New York State climatic conditions. Sci. Hortic. 227, 22–37 (2018).
Virlet, N., Costes, E., Martinez, S., Kelner, J.-J. & Regnard, J.-L. Multispectral airborne imagery in the field reveals genetic determinisms of morphological and transpiration traits of an apple tree hybrid population in response to water deficit. J. Exp. Bot. 66, 5453–5465 (2015).
Wang, H. et al. Mapping QTLs for water-use efficiency reveals the potential candidate genes involved in regulating the trait in apple under drought stress. BMC Plant Biol. 18, 136 (2018).
Liu, B., Cheng, L., Ma, F., Zou, Y. & Liang, D. Growth, biomass allocation, and water use efficiency of 31 apple cultivars grown under two water regimes. Agrofor. Syst. 84, 117–129 (2012).
Zhou, S. et al. Comprehensive genomic analysis and expression profiling of Argonaute gene family and examination of their regulatory roles in water-use efficiency and abiotic stress responses in apple. Acta Physiol. Plant. 38, 1–14 (2016).
Dong, Q. et al. Genome-wide analyses of genes encoding FK506-binding proteins reveal their involvement in abiotic stress responses in apple. BMC Genom. 19, 707 (2018).
Wang, N. et al. Functions of two Malus hupehensis (Pamp.) Rehd. YTPs (MhYTP1 and MhYTP2) in biotic- and abiotic-stress responses. Plant Sci. 261, 18–27 (2017).
Wang, N., Yue, Z., Liang, D. & Ma, F. Genome-wide identification of members in the YTH domain-containing RNA-binding protein family in apple and expression analysis of their responsiveness to senescence and abiotic stresses. Gene 538, 292–305 (2014).
Liu, C. et al. Overexpression of MhYTP2 enhances apple water-use efficiency by activating ABA and ethylene signaling. Environ. Exp. Bot. 157, 260–268 (2019).
Guo, T. et al. Overexpression of the RNA binding protein MhYTP1 in transgenic apple enhances drought tolerance and WUE by improving ABA level under drought condition. Plant Sci. 280, 397–407 (2019).
Fazio, G., Chang, L., Grusak, M. A. & Robinson, T. L. Apple rootstocks influence mineral nutrient concentration of leaves and fruit. N. Y. Fruit Q 23, 11–15 (2015).
Fazio, G., Lordan, J., Francescatto, P. & Robinson, T. L. Breeding apple rootstocks to match cultural and nutrient requirements of scionvarieties. FRUIT Q 26, 25–30 (2018).
Lordan, J., Fazio, G., Francescatto, P. & Robinson, T. Effects of apple (Malus×domestica) rootstocks on scion performance and hormone concentration. Sci. Hortic. 225, 96–105 (2017).
Neilsen, G. H., Neilsen, D., Guak, S. & Forge, T. The effect of deficit irrigation and crop load on leaf and fruit nutrition of fertigated ‘Ambrosia’/‘M.9’ apple. HortScience 50, 1387–1393 (2015).
Fazio, G., Kviklys, D., Grusak, M. A. & Robinson, T. Phenotypic diversity and QTL mapping of absorption and translocation of nutrients by apple rootstocks. Asp. Appl. Biol. 119, 37–50 (2013).
Scheper, R. & Fisher, B. Comparing methods to determine European canker resistance in apple tree accessions. N. Z. Plant Prot. 63, 280 (2010).
Porto, D. D. et al. Transcription profiling of the chilling requirement for bud break in apples: a putative role for FLC-like genes. J. Exp. Bot. 66, 2659–2672 (2015).
Porto, D. D. et al. Structural genomics and transcriptional characterization of the dormancy-associated MADS-box genes during bud dormancy progression in apple. Tree Genet. Genomes 12, 46 (2016).
Kumar, G. et al. Comparative phylogenetic analysis and transcriptional profiling of MADS-box gene family identified DAM and FLC-like genes in apple (Malus × domestica). Sci. Rep. 6, 20695 (2016).
Kumar, G. et al. Chilling affects phytohormone and post-embryonic development pathways during bud break and fruit set in apple (Malus domestica Borkh.). Sci. Rep. 7, 42593 (2017).
Takeuchi, T. et al. RNA-sequencing analysis identifies genes associated with chilling-mediated endodormancy release in apple. J. Am. Soc. Hortic. Sci. 143, 194–206 (2018).
Falavigna, V., da, S., Guitton, B., Costes, E. & Andrés, F. I want to (bud) break free: the potential role of DAM and SVP-Like genes in regulating dormancy cycle in temperate fruit trees. Front. Plant Sci. 9, 1990 (2019).
Saito, T. et al. Lipid droplet-associated gene expression and chromatin remodelling in LIPASE 5’-upstream region from beginning- to mid-endodormant bud in ‘Fuji’ apple. Plant Mol. Biol. 95, 441–449 (2017).
Falavigna, V. et al. Evolutionary diversification of galactinol synthases in Rosaceae: adaptive roles of galactinol and raffinose during apple bud dormancy. J. Exp. Bot. 69, 1247–1259 (2018).
Tarancón, C., González-Grandío, E., Oliveros, J. C., Nicolas, M. & Cubas, P. A conserved carbon starvation response underlies bud dormancy in woody and herbaceous species. Front. Plant Sci. 8, 788 (2017).
van Dyk, M. M., Soeker, M. K., Labuschagne, I. F. & Rees, D. J. G. Identification of a major QTL for time of initial vegetative budbreak in apple (Malus × domestica Borkh.). Tree Genet. Genomes 6, 489–502 (2010).
Celton, J.-M. et al. Deciphering the genetic determinism of bud phenology in apple progenies: a new insight into chilling and heat requirement effects on flowering dates and positional candidate genes. New Phytol. 192, 378–392 (2011).
Allard et al. Detecting QTLs and putative candidate genes involved in budbreak and flowering time in an apple multiparental population. J. Exp. Bot. 67, 2875–2888 (2016).
Kumar, G., Rattan, U. K. & Singh, A. K. Chilling-mediated DNA methylation changes during dormancy and its release reveal the importance of epigenetic regulation during winter dormancy in apple (Malus × domestica Borkh.). PLoS ONE 11, e0149934 (2016).
Wigge, P. A. et al. Integration of spatial and temporal information during floral induction in Arabidopsis. Science 309, 1056–1059 (2005).
Corbesier, L. et al. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 316, 1030–1033 (2007).
Jaeger, K. E. & Wigge, P. A. FT protein acts as a long-range signal in Arabidopsis. Curr. Biol. 17, 1050–1054 (2007).
Hättasch, C., Flachowsky, H., Kapturska, D. & Hanke, M.-V. Isolation of flowering genes and seasonal changes in their transcript levels related to flower induction and initiation in apple (Malus domestica). Tree Physiol. 28, 1459–1466 (2008).
Kotoda, N. et al. Molecular characterization of FLOWERING LOCUS T-like genes of apple (Malus × domestica Borkh.). Plant Cell Physiol. 51, 561–575 (2010).
Wilkie, J. D., Sedgley, M. & Olesen, T. Regulation of floral initiation in horticultural trees. J. Exp. Bot. 59, 3215–3228 (2008).
Mutasa-Göttgens, E. & Hedden, P. Gibberellin as a factor in floral regulatory networks. J. Exp. Bot. 60, 1979–1989 (2009).
Haberman, A. et al. Different flowering response to various fruit loads in apple cultivars correlates with degree of transcript reaccumulation of a TFL1-encoding gene. Plant J. Cell Mol. Biol. 87, 161–173 (2016).
Kotoda, N. & Wada, M. MdTFL1, a TFL1-like gene of apple, retards the transition from the vegetative to reproductive phase in transgenic Arabidopsis. Plant Sci. 168, 95–104 (2005).
Guitton, B. et al. Genetic control of biennial bearing in apple. J. Exp. Bot. 63, 131–149 (2012).
Guitton, B. et al. Analysis of transcripts differentially expressed between fruited and deflowered ‘Gala’ adult trees: a contribution to biennial bearing understanding in apple. BMC Plant Biol. 16, 55 (2016).
van Nocker, S. & Gardiner, S. E. Breeding better cultivars, faster: applications of new technologies for the rapid deployment of superior horticultural tree crops. Hortic. Res. 1, 14022 (2014).
Wang, J.-W. et al. MiRNA control of vegetative phase change in trees. PLoS Genet. 7, e1002012 (2011).
Xing, L. et al. Genome-wide identification of vegetative phase transition-associated microRNAs and target predictions using degradome sequencing in Malus hupehensis. BMC Genom. 15, 1125 (2014).
Giovannoni, J. J. Genetic regulation of fruit development and ripening. Plant Cell 16, S170–S180 (2004).
Yang, S. F. & Hoffman, N. E. Ethylene biosynthesis and its regulation in higher plants. Annu. Rev. Plant Physiol. 35, 155–189 (1984).
Alexander, L. & Grierson, D. Ethylene biosynthesis and action in tomato: a model for climacteric fruit ripening. J. Exp. Bot. 53, 2039–2055 (2002).
Bleecker, A. Ethylene perception and signaling: an evolutionary perspective. Trends. Plant Sci. 4, 269–274 (1999).
Binder, B. M. The ethylene receptors: complex perception for a simple gas. Plant Sci. 175, 8–17 (2008).
Tadiello, A. et al. Interference with ethylene perception at receptor level sheds light on auxin and transcriptional circuits associated with the climacteric ripening of apple fruit (Malus × domestica Borkh.). Plant J. Cell Mol. Biol. 88, 963–975 (2016).
Busatto, N., Tadiello, A., Trainotti, L. & Costa, F. Climacteric ripening of apple fruit is regulated by transcriptional circuits stimulated by cross-talks between ethylene and auxin. Plant Signal. Behav. 12, 2194–2202 (2017).
Busatto, N. et al. Apple fruit superficial scald resistance mediated by ethylene inhibition is associated with diverse metabolic processes. Plant J. 93, 270–285 (2018).
Celton, J.-M. et al. Widespread anti-sense transcription in apple is correlated with siRNA production and indicates a large potential for transcriptional and/or post-transcriptional control. New Phytol. 203, 287–299 (2014).
Segonne, S. M. et al. Multiscale investigation of mealiness in apple: an atypical role for a pectin methylesterase during fruit maturation. BMC Plant Biol. 14, 375 (2014).
Dheilly, E. et al. Cell wall dynamics during apple development and storage involves hemicellulose modifications and related expressed genes. BMC Plant Biol. 16, 201 (2016).
Farneti, B. et al. Genome-wide association study unravels the genetic control of the apple volatilome and its interplay with fruit texture. J. Exp. Bot. 68, 1467–1478 (2017).
Iglesias, I., Echeverría, G. & Soria, Y. Differences in fruit colour development, anthocyanin content, fruit quality and consumer acceptability of eight ‘Gala’ apple strains. Sci. Hortic. 119, 32–40 (2008).
White, A. G. & Lespinasse, Y. The inheritance of fruit colour in apple (Malus pumila Mill.). Agronomie 6, 105–108 (1986).
Cheng, F. S., Weeden, N. F. & Brown, S. K. Identification of co-dominant RAPD markers tightly linked to fruit skin color in apple. Theor. Appl. Genet. 93, 222–227 (1996).
Chagné, D. et al. Mapping a candidate gene (MdMYB10) for red flesh and foliage colour in apple. BMC Genom. 8, 212 (2007).
Espley, R. V. et al. Multiple repeats of a promoter segment causes transcription factor autoregulation in red apples. Plant Cell 21, 168–183 (2009).
Takos, A. M. et al. Light-induced expression of a MYB gene regulates anthocyanin biosynthesis in red apples. Plant Physiol. 142, 1216–1232 (2006).
Lin-Wang, K. et al. High temperature reduces apple fruit colour via modulation of the anthocyanin regulatory complex. Plant Cell Environ. 34, 1176–1190 (2011).
Chagné, D. et al. QTL and candidate gene mapping for polyphenolic composition in apple fruit. BMC Plant Biol. 12, 12 (2012).
Kumar, S. et al. Genomic selection for fruit quality traits in apple (Malus × domestica Borkh.). PLoS ONE 7, e36674 (2012).
Lozano, L. et al. Feasibility of genome-wide association analysis using a small single nucleotide polymorphismpanel in an apple breeding population segregating for fruit skin color. J. Am. Soc. Hortic. Sci. 139, 619–626 (2014).
Sooriyapathirana, S. S. et al. QTL analysis and candidate gene mapping for skin and flesh color in sweet cherry fruit (Prunus avium L.). Tree Genet. Genomes 6, 821–832 (2010).
Chagné, D. et al. A functional genetic marker for apple red skin coloration across different environments. Tree Genet. Genomes 12, 67 (2016).
Telias, A. et al. Apple skin patterning is associated with differential expression of MYB10. BMC Plant Biol. 11, 93 (2011).
Zhang, Y., Li, P. & Cheng, L. Developmental changes of carbohydrates, organic acids, amino acids, and phenolic compounds in ‘Honeycrisp’ apple flesh. Food Chem. 123, 1013–1018 (2010).
Ma, B. et al. Comparative assessment of sugar and malic acid composition in cultivated and wild apples. Food Chem. 172, 86–91 (2015).
Berüter, J. Carbohydrate metabolism in two apple genotypes that differ in malate accumulation. J. Plant. Physiol. 161, 1011–1029 (2004).
Yao, Y.-X. et al. Molecular cloning of three malic acid related genes MdPEPC, MdVHA-A, MdcyME and their expression analysis in apple fruits. Sci. Hortic. 122, 404–408 (2009).
Yao, Y.-X., Li, M., Zhai, H., You, C.-X. & Hao, Y.-J. Isolation and characterization of an apple cytosolic malate dehydrogenase gene reveal its function in malate synthesis. J. Plant. Physiol. 168, 474–480 (2011).
Maliepaard, C. et al. Aligning male and female linkage maps of apple (Malus pumila Mill.) using multi-allelic markers. Theor. Appl. Genet. 97, 60–73 (1998).
Kenis, K., Keulemans, J. & Davey, M. W. Identification and stability of QTLs for fruit quality traits in apple. Tree Genet. Genomes 4, 647–661 (2008).
Khan, S. A. et al. Differences in acidity of apples are probably mainly caused by a malic acid transporter gene on LG16. Tree Genet. Genomes 9, 475–487 (2013).
Xu, K., Wang, A. & Brown, S. Genetic characterization of the Ma locus with pH and titratable acidity in apple. Mol. Breed. 30, 899–912 (2012).
Ma, B. et al. Genes encoding aluminum-activated malate transporter II and their association with fruit acidity in apple. Plant Genome 8, 3 (2015).
Zhang, Q. et al. Identification, characterization, and utilization of genome-wide simple sequence repeats to identify a QTL for acidity in apple. BMC Genom. 13, 537 (2012).
Sun, R. et al. A dense SNP genetic map constructed using restriction site-associated DNA sequencing enables detection of QTLs controlling apple fruit quality. BMC Genom. 16, 747 (2015).
Verma, S. et al. Two large-effect QTLs, Ma and Ma3, determine genetic potential for acidity in apple fruit: breeding insights from a multi-family study. Tree Genet. Genomes 15, 18 (2019).
Jia, D. et al. Apple fruit acidity is genetically diversified by natural variations in three hierarchical epistatic genes: MdSAUR37, MdPP2CH and MdALMTII. Plant J. Cell Mol. Biol. 95, 427–443 (2018).
Bai, Y., Dougherty, L., Cheng, L. & Xu, K. A co-expression gene network associated with developmental regulation of apple fruit acidity. Mol. Genet. Genom. 290, 1247–1263 (2015).
Bai, Y., Dougherty, L., Cheng, L., Zhong, G.-Y. & Xu, K. Uncovering co-expression gene network modules regulating fruit acidity in diverse apples. BMC Genom. 16, 612 (2015).
Hu, D.-G., Sun, C.-H., Sun, M.-H. & Hao, Y.-J. MdSOS2L1 phosphorylates MdVHA-B1 to modulate malate accumulation in response to salinity in apple. Plant Cell Rep. 35, 705–718 (2016).
Hu, D.-G. et al. The R2R3-MYB transcription factor MdMYB73 is involved in malate accumulation and vacuolar acidification in apple. Plant J. Cell Mol. Biol. 91, 443–454 (2017).
Giovannoni, J. J., Wing, R. A., Ganal, M. W. & Tanksley, S. D. Isolation of molecular markers from specific chromosomal intervals using DNA pools from existing mapping populations. Nucleic Acids Res. 19, 6553–6558 (1991).
Gianfranceschi, L., Seglias, N., Tarchini, R., Komjanc, M. & Gessler, C. Simple sequence repeats for the genetic analysis of apple. Theor. Appl. Genet. 96, 1069–1076 (1998).
Liebhard, R. et al. Development and characterisation of 140 new microsatellites in apple (Malus × domestica Borkh.). Mol. Breed. 10, 217–241 (2002).
Silfverberg-Dilworth, E. et al. Microsatellite markers spanning the apple (Malus × domestica Borkh.) genome. Tree Genet. Genomes 2, 202–224 (2006).
Bus, V. G. M. et al. The Vh2 and Vh4 scab resistance genes in two differential hosts derived from Russian apple R12740-7A map to the same linkage group of apple. Mol. Breed. 15, 103–116 (2005).
Patocchi, A. et al. Identification by genome scanning approach (GSA) of a microsatellite tightly associated with the apple scab resistance gene Vm. Genome 48, 630–636 (2005).
Gygax, M. et al. Molecular markers linked to the apple scab resistance gene Vbj derived from Malus baccata jackii. Theor. Appl. Genet. 109, 1702–1709 (2004).
Erdin, N. et al. Mapping of the apple scab-resistance gene Vb. Genome 49, 1238–1245 (2006).
Patocchi, A., Bigler, B., Koller, B., Kellerhals, M. & Gessler, C. Vr2: a new apple scab resistance gene. Theor. Appl. Genet. 109, 1087–1092 (2004).
Durel, C.-E., Denancé, C. & Brisset, M.-N. Two distinct major QTL for resistance to fire blight co-localize on linkage group 12 in apple genotypes ‘Evereste’ and Malus floribunda clone 821. Genome 52, 139–147 (2009).
Peil, A. et al. Strong evidence for a fire blight resistance gene of Malus robusta located on linkage group 3. Plant Breed. 126, 470–475 (2007).
Gardiner, S. E. et al. Putative resistance gene markers associated with quantitative trait loci for fire blight resistance in Malus ‘Robusta 5’ accessions. BMC Genet. 13, 25 (2012).
Evans, K. M. & James, C. M. Identification of SCAR markers linked to Pl-w mildew resistance in apple. Theor. Appl. Genet. 106, 1178–1183 (2003).
Dunemann, F., Peil, A., Urbanietz, A. & Garcia‐Libreros, T. Mapping of the apple powdery mildew resistance gene Pl1 and its genetic association with an NBS-LRR candidate resistance gene. Plant Breed. 126, 476–481 (2007).
Calenge, F. et al. Quantitative trait loci (QTL) analysis reveals both broad-spectrum and isolate-specific QTL for scab resistance in an apple progeny challenged with eight isolates of Venturia Inaequalis. Phytopathology 94, 370–379 (2004).
Calenge, F. & Durel, C.-E. Both stable and unstable QTLs for resistance to powdery mildew are detected in apple after four years of field assessments. Mol. Breed. 17, 329–339 (2006).
Khan, M. A., Duffy, B., Gessler, C. & Patocchi, A. QTL mapping of fire blight resistance in apple. Mol. Breed. 17, 299–306 (2006).
Galli, P., Patocchi, A., Broggini, G. A. L. & Gessler, C. The Rvi15 (Vr2) apple scab resistance locus contains three TIR-NBS-LRR genes. Mol. Plant Microbe Interact. 23, 608–617 (2010).
Parravicini, G. et al. Identification of serine/threonine kinase and nucleotide-binding site-leucine-rich repeat (NBS-LRR) genes in the fire blight resistance quantitative trait locus of apple cultivar ‘Evereste’. Mol. Plant Pathol. 12, 493–505 (2011).
Fahrentrapp, J. et al. A candidate gene for fire blight resistance in Malus × robusta 5 is coding for a CC–NBS–LRR. Tree Genet. Genomes 9, 237–251 (2013).
Patocchi, A., Gianfranceschi, L. & Gessler, C. Towards the map-based cloning of Vf: fine and physical mapping of the Vf region. Theor. Appl. Genet. 99, 1012–1017 (1999).
Soriano, J. M. et al. Fine mapping of the gene Rvi18 (V25) for broad-spectrum resistance to apple scab, and development of a linked SSR marker suitable for marker-assisted breeding. Mol. Breed. 34, 2021–2032 (2014).
Cova, V. et al. High-resolution genetic and physical map of the Rvi1 (Vg) apple scab resistance locus. Mol. Breed. 35, 16 (2015).
Cova, V. et al. Fine mapping of the Rvi5 (Vm) apple scab resistance locus in the ‘Murray’ apple genotype. Mol. Breed. 35, 200 (2015).
Padmarasu, S. et al. Identification of a leucine-rich repeat receptor-like serine/threonine-protein kinase as a candidate gene for Rvi12 (Vb)-based apple scab resistance. Mol. Breed. 38, 73 (2018).
Emeriewen, O. F. et al. Towards map-based cloning of FB_Mfu10: identification of a receptor-like kinase candidate gene underlying the Malus fusca fire blight resistance locus on linkage group 10. Mol. Breed. 38, 106 (2018).
Wöhner, T. et al. Homologs of the FB_MR5 fire blight resistance gene of Malus × robusta 5 are present in other Malus wild species accessions. Tree Genet. Genomes 12, 2 (2016).
van de Weg, E. et al. Epistatic fire blight resistance QTL alleles in the apple cultivar ‘Enterprise’ and selection X-6398 discovered and characterized through pedigree-informed analysis. Mol. Breed. 38, 5 (2018).
Ellis, J., Dodds, P. & Pryor, T. Structure, function and evolution of plant disease resistance genes. Curr. Opin. Plant Biol. 3, 278–284 (2000).
Nishitani, C. et al. Efficient genome editing in apple using a CRISPR/Cas9 system. Sci. Rep. 6, 31481 (2016).
Charrier, A. et al. Efficient targeted mutagenesis in apple and first time edition of pear using the CRISPR-Cas9 system. Front. Plant Sci. 10, 40 (2019).
Pessina, S. et al. Characterization of the MLO gene family in Rosaceae and gene expression analysis in Malus domestica. BMC Genom. 15, 618 (2014).
Pessina, S. et al. Frequency of a natural truncated allele of MdMLO19 in the germplasm of Malus domestica. Mol. Breed. 37, 7 (2017).
Troggio, M. et al. Apple, from genome to breeding. Tree Genet. Genomes 8, 509–529 (2012).
Bastiaanse, H. et al. Scab resistance in ‘Geneva’ apple is conditioned by a resistance gene cluster with complex genetic control. Mol. Plant Pathol. 17, 159–172 (2016).
Jung, S. et al. The Genome Database for Rosaceae (GDR): year 10 update. Nucleic Acids Res. 42, D1237–D1244 (2014).
Gardiner, S. E. Novel genetic marker technologies revolutionize apple breeding©. Acta Hortic. 1174, 23–30 (2017).
Chagné D., et al. Validation of SNP markers for fruit quality and disease resistance loci in apple (Malus × domestica Borkh.) using the OpenArray® platform. Hortic. Res. 6, 30 (2019).
Vanderzande, S. et al. Crossing the finish line: how to develop diagnostic DNA tests as breeding tools after QTL discovery. J. Hortic. 05, 1–6 (2018).
Tartarini, S., Gianfranceschi, L., Sansavini, S. & Gessler, C. Development of reliable PCR markers for the selection of the Vf gene conferring scab resistance in apple. Plant Breed. 118, 183–186 (1999).
Bus, V. G. M. et al. Genome mapping of three major resistance genes to woolly apple aphid (Eriosoma lanigerum Hausm.). Tree Genet. Genomes 4, 223–236 (2008).
Bus V. G. M., Esmenjaud D., Buck E. & Laurens F. in Genetics and Genomics of Rosaceae (eds Folta K. M. & Gardiner S. E.) 563–599 (Springer, New York, NY, 2009).
Bus, V. et al. Progress in pipfruit resistance breeding and research at Plant & Food Research. Acta Hortic. 1172, 7–14 (2017).
Peace, C. P. DNA-informed breeding of rosaceous crops: promises, progress and prospects. Hortic. Res. 4, 17006 (2017).
Baumgartner, I. O. et al. Development of SNP-based assays for disease resistance and fruit quality traits in apple (Malus × domestica Borkh.) and validation in breeding pilot studies. Tree Genet. Genomes 12, 35 (2016).
Gardiner, S. et al. Fire blight resistance from Malus ‘Robusta 5’ - identification and application of genetic markers. I Internationl Apple Symposium (Yangling, 2016).
Bus, V. G. M. et al. Genetic mapping of the European canker (Neonectria ditissima) resistance locus Rnd1 from Malus ‘Robusta 5’. Tree Genet. Genomes 15, 25 (2019).
Bassett, H. et al. Marker assisted selection in an apple rootstock breeding family. Acta Hortic. 1100, 25–28 (2015).
Moriya, S. et al. Identification and genetic characterization of a quantitative trait locus for adventitious rooting from apple hardwood cuttings. Tree Genet. Genomes 11, 59 (2015).
Evans, K. & Peace, C. in Achieving Sustainable Cultivation of Apples (ed. Evans, K.) 189–216 (Burleigh Dodds Science Publishing, London, 2017).
Campbell, N. R., Harmon, S. A. & Narum, S. R. Genotyping-in-Thousands by sequencing (GT-seq): a cost effective SNP genotyping method based on custom amplicon sequencing. Mol. Ecol. Resour. 15, 855–867 (2015).
Browning, B. L. & Browning, S. R. Genotype imputation with millions of reference samples. Am. J. Hum. Genet. 98, 116–126 (2016).
Isik, F., Kumar, S., Martínez-García, P. J., Iwata, H. & Yamamoto, T. in Advances in Botanical Research (eds. Plomion, C. & Adam-Blondon, A.-F.). 93–124 (Academic Press, Cambridge, 2015).
Georges, M., Charlier, C. & Hayes, B. Harnessing genomic information for livestock improvement. Nat. Rev. Genet. 20, 135–156 (2019).
Voss-Fels, K. P., Cooper, M. & Hayes B. J. Accelerating crop genetic gains with genomic selection. Theor. Appl. Genet. 132, 669–686 (2018).
Muranty, H. et al. Accuracy and responses of genomic selection on key traits in apple breeding. Hortic. Res. 2, 15060 (2015).
Meuwissen, T. H., Hayes, B. J. & Goddard, M. E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157, 1819–1829 (2001).
Kumar, S. et al. Genome-enabled estimates of additive and nonadditive genetic variances and prediction of apple phenotypes across environments. G3 5, 2711–2718 (2015).
Migault, V., Pallas, B. & Costes, E. Combining genome-wide information with a functional structural plant model to simulate 1-year-old apple tree architecture. Front. Plant Sci. 7, 2065 (2017).
Crossa, J. et al. Genomic selection in plant breeding: methods, models, and perspectives. Trends. Plant Sci. 22, 961–975 (2017).
Hurlbert, S. H. Pseudoreplication and the design of ecological field experiments. Ecol. Monogr. 54, 187–211 (1984).
Rolf, M. M. et al. Impact of reduced marker set estimation of genomic relationship matrices on genomic selection for feed efficiency in Angus cattle. BMC Genet. 11, 24 (2010).
Migicovsky, Z. & Myles, S. Exploiting wild relatives for genomics-assisted breeding of perennial crops. Front. Plant Sci. 8, 460 (2017).
Rabier, C.-E., Barre, P., Asp, T., Charmet, G. & Mangin, B. On the accuracy of genomic selection. PLoS ONE 11, e0156086 (2016).
de los Campos, G., Hickey, J. M., Pong-Wong, R., Daetwyler, H. D. & Calus, M. P. L. Whole-genome regression and prediction methods applied to plant and animal breeding. Genetics 193, 327–345 (2013).
Heslot, N., Yang, H.-P., Sorrells, M. E. & Jannink, J.-L. Genomic selection in plant breeding: a comparison of models. Crop Sci. 52, 146–160 (2012).
Goddard, M. E. & Meuwissen, T. H. E. The use of linkage disequilibrium to map quantitative trait loci. Aust. J. Exp. Agric. 45, 837–845 (2005).
Wray, N. R., Goddard, M. E. & Visscher, P. M. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 17, 1520–1528 (2007).
Goddard, M. E., Wray, N. R., Verbyla, K. & Visscher, P. M. Estimating effects and making predictions from genome-wide marker data. Stat. Sci. 24, 517–529 (2009).
Piaskowski, J. et al. Genomic heritability estimates in sweet cherry reveal non-additive genetic variance is relevant for industry-prioritized traits. BMC Genet. 19, 23 (2018).
Hardner, C. M., Kumar, S., Peace, C. M., Luby, J. & Evans, K. M. Reconstructing relationship matrices from dense SNP arrays for the prediction of genetic potential in unreplicated multilocation plantings of apple progeny. Acta Hortic. 1127, 275–282 (2016).
Yue, C. et al. An investigation of U.S. apple producers’ trait prioritization—evidence from audience surveys. HortScience 48, 1378–1384 (2013).
Hickey, J. M. et al. Genomic prediction unifies animal and plant breeding programs to form platforms for biological discovery. Nat. Genet. 49, 1297–1303 (2017).
Iwata, H., Minamikawa, M. F., Kajiya-Kanegae, H., Ishimori, M. & Hayashi, T. Genomics-assisted breeding in fruit trees. Breed. Sci. 66, 100–115 (2016).
Covarrubias-Pazaran, G. et al. Multivariate GBLUP improves accuracy of genomic gelection for yield and fruit weight in biparental populations of Vaccinium macrocarpon Ait. Front. Plant Sci. 9, 1310 (2018).
Cooper, M., Gho, C., Leafgren, R., Tang, T. & Messina, C. Breeding drought-tolerant maize hybrids for the US corn-belt: discovery to product. J. Exp. Bot. 65, 6191–6204 (2014).
MacLeod, I. M. et al. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genom. 17, 144 (2016).
Stephens, J. C. et al. Haplotype variation and linkage disequilibrium in 313 human genes. Science 293, 489–493 (2001).
Qian, L. et al. Exploring and harnessing haplotype diversity to improve yield stability in crops. Front. Plant Sci. 8, 1534 (2017).
Werner, C. R. et al. Effective genomic selection in a narrow-genepool crop with low-density markers: Asian rapeseed as an example. Plant Genome 11, 170084 (2018).
Flachowsky, H. et al. Application of a high-speed breeding technology to apple (Malus × domestica) based on transgenic early flowering plants and marker-assisted selection. New Phytol. 192, 364–377 (2011).
Wenzel, S., Flachowsky, H. & Hanke, M.-V. The Fast-track breeding approach can be improved by heat-induced expression of the FLOWERING LOCUS T genes from poplar (Populus trichocarpa) in apple (Malus × domestica Borkh.). Plant Cell Tissue Organ Culture 115, 127–137 (2013).
Yamagishi, N., Kishigami, R. & Yoshikawa, N. Reduced generation time of apple seedlings to within a year by means of a plant virus vector: a new plant-breeding technique with no transmission of genetic modification to the next generation. Plant Biotechnol. J. 12, 60–68 (2014).
Schlathölter, I. et al. Generation of advanced fire blight-resistant apple (Malus × domestica) selections of the fifth generation within 7 years of applying the early flowering approach. Planta 247, 1475–1488 (2018).
Hardner, C. M. et al. Prediction of genetic value for sweet cherry fruit maturity among environments using a 6K SNP array. Hortic. Res. 6, 6 (2019).
Han, M. et al. Insertion of a solo LTR retrotransposon associates with spur mutations in ‘Red Delicious’ apple (Malus × domestica). Plant Cell Rep. 36, 1375–1385 (2017).
El-Sharkawy, I., Liang, D. & Xu, K. Transcriptome analysis of an apple (Malus × domestica) yellow fruit somatic mutation identifies a gene network module highly associated with anthocyanin and epigenetic regulation. J. Exp. Bot. 66, 7359–7376 (2015).
Thieme, M. et al. Inhibition of RNA polymerase II allows controlled mobilisation of retrotransposons for plant breeding. Genome Biol. 18, 134 (2017).
Jung, S. et al. 15 years of GDR: new data and functionality in the Genome Database for Rosaceae. Nucleic Acids Res. 47, D1137–D1145 (2019).
Ficklin, S. P. et al. Tripal: a construction toolkit for online genome databases. Database 2011, bar044 (2011).
Sanderson, L.-A. et al. Tripal v1.1: a standards-based toolkit for construction of online genetic and genomic databases. Database 2013, bat075 (2013).
Buels, R. et al. JBrowse: a dynamic web platform for genome visualization and analysis. Genome Biol. 17, 66 (2016).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Finn, R. D. et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 (2017).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000).
The Gene Ontology Consortium. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 45, D331–D338 (2017).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinform. 10, 421 (2009).
Van Ooijen, J. MapQTL® 6, Software for the Mapping of Quantitative Trait Loci in Experimental Populations of Diploid Species (Kyazma B.V., Wageningen, 2009).
Pacheco, I. et al. QTL mapping for brown rot (Monilinia fructigena) resistance in an intraspecific peach (Prunus persica L. Batsch) F1 progeny. Tree Genet. Genomes 10, 1223–1242 (2014).
Salvi, S. & Tuberosa, R. The crop QTLome comes of age. Curr. Opin. Biotechnol. 32, 179–185 (2015).
Peleman, J. D. & van der Voort, J. R. Breeding by Design. Trends Plant Sci. 8, 330–334 (2003).
Acknowledgements
The authors are grateful to the various funding agencies and projects that contributed to the presented work including, but not exclusively, the FruitBreedomics project no. 265582 “Integrated approach for increasing breeding efficiency in fruit tree crops” co-funded by the EU Seventh Framework Program, the USDA’s National Institute of Food and Agriculture (NIFA)–Specialty Crop Research Initiative funded projects “RosBREED: Enabling marker-assisted breeding in Rosaceae” (2009-51181-05808), “RosBREED: Combining disease resistance with horticultural quality in new rosaceous cultivars” (2014-51181-22378), and “Genome Database for Rosaceae: Empowering specialty crop research through big-data driven discovery and application in breeding (2014-51181-2237), NIFA’s National Research Support Program 10 “Database resources for crop genomics, genetics and breeding research”, the IDEEV, Laboratoire d’Excellence BASC, and the Inserm ATIP-Avenir program. Research undertaken in New Zealand was supported by Prevar™ and the Strategic Science Investment Fund from the New Zealand Ministry of Business, Innovation and Employment. ZM was supported by NSF 1546869. CH’s contribution was funded through The Queensland Alliance for Agriculture and Food Innovation, research institute of the University of Queensland supported by the Queensland Government.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peace, C.P., Bianco, L., Troggio, M. et al. Apple whole genome sequences: recent advances and new prospects. Hortic Res 6, 59 (2019). https://doi.org/10.1038/s41438-019-0141-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-019-0141-7
This article is cited by
-
Forgotten forest relics: Apple trees (Malus spp.) in eastern U.S. forests
Biodiversity and Conservation (2023)
-
Breeding and genetics of disease resistance in temperate fruit trees: challenges and new opportunities
Theoretical and Applied Genetics (2022)
-
Transcriptome analysis reveals the effects of strigolactone on shoot regeneration of apple
Plant Cell Reports (2022)
-
Integration of Infinium and Axiom SNP array data in the outcrossing species Malus × domestica and causes for seemingly incompatible calls
BMC Genomics (2021)
-
Tracing founder haplotypes of Japanese apple varieties: application in genomic prediction and genome-wide association study
Horticulture Research (2021)
