
Abstract
Purpose
To develop an evidence-based clinical practice guideline for the use of exome and genome sequencing (ES/GS) in the care of pediatric patients with one or more congenital anomalies (CA) with onset prior to age 1 year or developmental delay (DD) or intellectual disability (ID) with onset prior to age 18 years.
Methods
The Pediatric Exome/Genome Sequencing Evidence-Based Guideline Work Group (n = 10) used the Grading of Recommendations Assessment, Development and Evaluation (GRADE) evidence to decision (EtD) framework based on the recent American College of Medical Genetics and Genomics (ACMG) systematic review, and an Ontario Health Technology Assessment to develop and present evidence summaries and health-care recommendations. The document underwent extensive internal and external peer review, and public comment, before approval by the ACMG Board of Directors.
Results
The literature supports the clinical utility and desirable effects of ES/GS on active and long-term clinical management of patients with CA/DD/ID, and on family-focused and reproductive outcomes with relatively few harms. Compared with standard genetic testing, ES/GS has a higher diagnostic yield and may be more cost-effective when ordered early in the diagnostic evaluation.
Conclusion
We strongly recommend that ES/GS be considered as a first- or second-tier test for patients with CA/DD/ID.
Similar content being viewed by others

INTRODUCTION
Congenital anomalies (CA), developmental delay (DD), and intellectual disability (ID) are among the most common indications for genetic referral in the pediatric population and comprise a heterogeneous group of conditions that can impact a child’s physical, learning, or behavioral function. In contrast to early childhood mortality, which declined by 50% from 1990 to 2016, the prevalence of developmental disabilities was unchanged over the same period, according to the Global Burden of Diseases, Injuries and Risk Factors Study.1 This study also reported on the worldwide prevalence and years lived with disability for six developmental disabilities: ID, epilepsy, autism spectrum disorder, attention deficit–hyperactivity disorder, and hearing and vision loss among children younger than 5 years of age in 195 countries and territories. In 2016, the global prevalence of ID was 12.5 million (confidence interval [CI] 10.2–15.1 million) or 1,983 cases per 100,000 children (CI 1,611–2,397 per 100,000). The European Surveillance of Congenital Anomalies (EUROCAT) recorded a total prevalence of major congenital anomalies of 23.9 per 1,000 births from 2003 to 2007 with 80% of these livebirths, with congenital heart defects the most common nonchromosomal subgroup, at 6.5 per 1,000 births, followed by limb defects (3.8 per 1,000), urinary tract anomalies (3.1 per 1,000), and nervous system anomalies (2.3 per 1,000).2
Identification of an underlying diagnosis for CA/DD/ID can lead to changes in management that will influence mortality, morbidity, and reduce the burden on patients and families searching for answers (sometimes referred to as the “diagnostic odyssey”).
The increased use of exome and genome sequencing in the past decade has significantly changed clinical genetics practice, as well as medicine more broadly. The clinical application of next-generation sequencing–based assays is one of the long-term goals envisioned from the Human Genome Project completed in 2003. Advances continue, but the benefits have been gradual due to technical, logistic, and financial constraints. Recognizing the importance of these approaches and the nature of gradual change in 2017 the American College of Medical Genetics and Genomics (ACMG) initiated a systematic evidence-based review for the use and outcomes from exome and genome sequencing for pediatric patients with congenital anomalies presenting before one year of age or ID presenting before 18 years of age. Congenital anomalies are structural or functional abnormalities usually evident at birth, or shortly thereafter, and can be consequential to an individual’s life expectancy, health status, and physical or social functioning, and typically require medical intervention. Published in 2020, this review allows for development of the present evidence-based guideline to assess the clinical utility, value to stakeholders, and feasibility of exome/genome sequencing (ES/GS) going forward.3
The increased use of ES/GS has uncovered the broader spectrum of disease associated with genetic variants and further increased diagnostic yield leading to improved patient outcomes. ES/GS has also furthered the understanding of both the natural history of many disorders and expanded potential treatments; importantly, the utility of the technologies will continue to grow and include improvements in gene therapy and potential for gene editing. A molecular diagnosis allows patients with a rare disorder and their families to tap into a worldwide support network for their condition.
To promote high quality care, evidence-based guidelines based on the 2011 Institute of Medicine (IOM) standards are developed by transparently combining systematic review conclusions with other strong related evidence, principles of care, and inferences.4 Developing evidence-based guidelines for rare diseases can be challenging for several reasons. The heterogeneity of genetic diagnoses leads to variation in timing of diagnosis/intervention and affects whether an intervention is readily available. Many genetic disorders are not amenable to randomized clinical trials (for neither diagnosis nor treatment) because of their rarity and lack of equivalent comparison. The nature of publications as either case reports or case series with limited long-term outcomes also makes evidence gathering problematic. Nonetheless, establishing evidence-based clinical practice guidelines is critical to moving the field forward. Clinical guidelines are not meant to supersede clinical judgment but rather provide evidentiary basis for these judgments and potentially lead to uniform use and coverage.
Current state
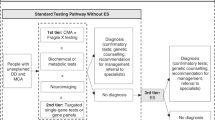
There is considerable variability in the use of molecular diagnostic tests in patients with CA/DD/ID. In 2010, the ACMG established chromosomal microarray (CMA) studies as a first-line genetic test in the consideration of a genetic diagnosis in children with CA and ID/DD.5 This recommendation does include children with a diagnosis of autism without other delays. Currently, practice options for second-tier testing varies depending on a number of factors, including regional/health-care system practice, insurance status, and provider/parental preference. One alternative to exome testing often considered is “panel testing” for a specific phenotype. These panels may be performed on an exome or genome platform with reports limited to the indicated genes/phenotype.
ES is available widely as a clinical tool with a number of commercial and academic laboratories offering this testing. Best practice includes familial comparators (“trio”) if available to help contextualize rare variants, but also can be effectively performed as proband only or duo, with diagnostic yield being slightly reduced compared with trio testing. More extensive familial testing may also be done to help determine significance of findings. Informed consent is required and should include a discussion of the possibility of identifying secondary findings (those variants that might be deemed medically actionable even if not directly related to the proband’s current condition) in the proband and potentially a family member. The ACMG has established a list of secondary findings analysis as an option and the utility of secondary findings has been established elsewhere.6,7,8 The limitations of ES in identifying genomic variants should be understood by the ordering clinician. ES typically does not detect intronic variants (unless immediately flanking a targeted exon). It also does not detect trinucleotide repeat expansions, methylation abnormalities, and may have only limited detection of copy-number variants.
GS is currently the first test of choice for a small number of clinics across the country, due in part to a limited performing lab option at this time. GS provides coverage of both array and exome targets, and further coverage of nonexome regions of the genome. We anticipate that the number of labs offering genomes will grow. Given the rapid changes anticipated, both ES and GS were included in the systematic review and these guidelines.
Current practice of ordering providers is also variable. There is a shortage of clinical genetics professionals across the United States, including physician-geneticists, pediatric genetic counselors, molecular/cytogeneticists, and lab services. The creation of guidelines will assist all clinicians (both genetics professionals and nongenetic professionals) to appropriately use and interpret ES/GS. Pretest genetic counseling including expectations of results, discussion of optional choices such as secondary findings and carrier status and follow-up plan remains standard of care. Pretest counseling also includes discussion of realistic expectations and potential benefits/harms in a nondirective manner, which genetic counselors are qualified and trained to do.
MATERIALS AND METHODS
Guideline panel composition
In 2020, the Pediatric Exome Sequencing/Genome Sequencing Guideline Work Group (Peds ES/GS GWG) was convened to develop an evidence-based guideline for the clinical use of ES/GS in patients with CA/DD/ID. Workgroup participants included American Board of Medical Genetics and Genomics certified members specializing in pediatric/adult clinical genetics and clinical laboratory genetics (L.A.D., F.M.H., K.M., L.J.M., H.M.K.), American Board of Psychiatry and Neurology certified pediatric and adult neurologists (T.W.Y., F.M.H.), an American Board of Genetic Counseling certified genetic counselor (S.B.), methodologists (M.R.M., J.M.), and a parent whose children have undergone ES and GS (D.M.). None of the working group members have any conflicts of interest according to ACMG board policy and independent review by the ACMG Conflict of Interest Committee.
To address the overarching research question, “Should exome sequencing or genome sequencing be used in the evaluation of patients with more than one congenital anomaly apparent before one year of age OR in patients with developmental disability/intellectual disability diagnosed prior to 18 years of age compared to standard testing without exome or genome sequencing?” the authors assessed evidence provided by a systematic review and a health technology assessment to develop recommendations about the appropriate use of ES/GS.3,9
Systematic review
The Grading of Recommendations Assessment, Development, and Evaluation (GRADE) Evidence to Decision (EtD) framework was used to develop and present evidence summaries and health-care recommendations.10 There were 167 studies included in the systematic evidence review (SER), 36 of which had a patient population greater than 20; additionally, smaller studies were included if they addressed specific outcomes.5,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70 The outcomes in the systematic review included (1) active clinical management (changes in medication, procedures and/or treatment); (2) monitoring and long-term clinical management (diagnostic testing, surveillance, referral to specialists or subspecialists, participation in a clinical trial, social services, and changes to lifestyle (e.g., diet); (3) family-focused outcomes (cascade genetic testing, referral to specialists, or changes in clinical management resulting from the diagnosis of a previously unknown disorder to family members of the index case); and (4) reproductive-focused outcomes (decisions to become pregnant, terminate a pregnancy, use assisted reproductive technologies, preimplantation genetic diagnosis, or donor sperm/egg, and to undergo previously unplanned additional prenatal testing such as chorionic villus sampling [CVS] or amniocentesis). Diagnostic yield and comparative diagnostic yield for both ES and GS were reported in the Ontario Health Technology Assessment (HTA).9 Behavioral/psychosocial outcomes and harms were considered by the systematic review, but so few were identified that they were not included in the summary of findings table; they were, however, considered in the EtD framework. The overall quality for each outcome was assessed using the GRADE methodology.71 The initial quality assessment corresponds to the study design, i.e., “high” for experimental studies (e.g., randomized clinical trials) and “low” for observational studies (e.g., cohort studies). GRADE considers five factors that might downgrade the study quality: limitations in study design (which pose risk of bias), inconsistency, indirectness, imprecision, and publication bias. Three factors can upgrade the quality of evidence: magnitude of effect, dose–response gradient, and, occasionally, types of residual confounding that could plausibly lead to an underestimation of the true effect of the intervention. The overall certainty of evidence was rated using a four-tiered (high, moderate, low, and very low) system.
GRADEpro software (https://www.gradepro.org) was used to create evidence profiles for the outcomes of interest.72,73 Outcomes were rated for practical and clinical importance by all members of the guideline panel (n = 8), from 1 (not critical to making a decision regarding the optimal patient care strategy) to 9 (critical to making a decision regarding optimal patient care). The final rating scores were reached by consensus during videoconferences with all guideline panel members. The EtD framework explicitly considered all four central domains (certainty of evidence, balance of benefits to harms, patients’ values and preferences, and resource utilization) for moving from evidence to recommendations.10,73 Each panel member voted on all 12 assessment questions under the four central domains via GRADEpro’s PanelVoice feature; consensus was reached by voting during videoconference calls.
Statistical analysis
Meta-analyses for each outcome from the systematic review described above were performed using a random-effects model, given the anticipated variation between studies, and were done in R (version 4.0.3) “meta” package. Single proportions were analyzed using generalized linear mixed models (GLMM; recommended for meta-analysis of single proportions, and 95% confidence intervals [CIs] were calculated for individual study results using the exact binomial interval (Clopper–Pearson interval [default]).74,75 The pooled results were summarized in forest plots. P < 0.05 (2-sided) was considered statistically significant.
RESULTS
Recommendation
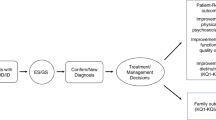
We strongly recommend ES and GS as a first-tier or second-tier test (guided by clinical judgment and often clinician–patient/family shared decision making after CMA or focused testing) for patients with one or more CAs prior to one year of age or for patients with DD/ID with onset prior to 18 years of age.
Justifications
Overall justification
The growing body of literature provides justification for a strong recommendation based on the balance of effects favoring the intervention (ES/GS) and desirable effects being potentially large with limited harms identified. The various stakeholders (i.e., health-care providers, patients, families, laboratories) are uniformly in favor of the use of ES/GS in obtaining a clinical diagnosis.
Detailed justification/summary of judgments
Problem
ES and GS are well-established diagnostic genetic testing approaches for identifying a genetic etiology among individuals with CA, DD, or ID. Clinical genetic testing by ES/GS can assist clinicians in confirming or establishing a clinical diagnosis that may lead to changes in management, obviate the need for further testing, and/or end the diagnostic odyssey. This may improve outcomes for the patient and family. However ES/GS is not always used nor available.
Desirable effects
Clinical utility includes short-term active clinical management changes (modifications to medications, procedures, or treatment) and long-term clinical management (referral to specialists, surveillance, or lifestyle changes).
Short-term active clinical management
A meta-analysis of data from 25 studies included in the ACMG SER reported that the rate of short-term clinical management impact was 8% (95% CI 6, 11) for all patients receiving ES/GS including those with no diagnostic finding (Fig. 1).11,12,13,14,15,16,17,18,19,20,21,22,24,25,26,27,28,29,30,31,32,33,34,35,36 This estimate combined ES and GS. The Ontario HTA reported a rate of 5.9% for ES and 10.4% for GS, with an overall rate of 6.3%.

CI confidence interval.
Long-term clinical management
A separate meta-analysis that included 19 studies from the ACMG SER (Fig. 2) showed a rate in long-term clinical management change of 10% (95% CI 7, 15) in all patients receiving ES/GS.19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37 The Ontario HTA reported a rate of 17.2% for ES and 20.8% for GS, with an overall rate of 17.5%.

CI confidence interval.
Reproductive-focused outcomes
Reproductive-focused outcomes were defined as decisions to become pregnant, terminate a pregnancy, use assisted reproductive technologies, use preimplantation genetic diagnosis, use donor sperm/egg, or undergo previously unplanned additional prenatal testing such as CVS or amniocentesis. Eight studies were included in a meta-analysis from the ACMG SER (Fig. 3) with a rate of 9% (95% CI 4, 21) for these outcomes in all patients receiving ES/GS.11,12,13,14,15,16,17,18

CI confidence interval.
Family-focused outcomes
Family-focused outcomes were defined as having an impact on family members of the patient, such as cascade genetic testing, referral to specialists, or changes in clinical management resulting from the diagnosis of a previously unknown disorder. Five studies were included in a meta-analysis from the ACMG SER (Fig. 4) with 4% (95% CI 2, 9) having this impact, again in all those receiving ES/GS.24,25,26,27,28

CI confidence interval.
Diagnostic yield
Diagnostic yield was outside the scope of the ACMG SER, although some information was gathered as part of the process. The Ontario HTA provided several types of estimates for diagnostic yield. The highest quality of evidence was for a direct comparison of the diagnostic yield of genome-wide sequencing (the terminology used in the HTA for ES/GS) versus standard genetic testing. The level of detail describing standard genetic testing was inconsistently reported, but typically it included CMA, candidate single-gene testing, or large gene panel testing. This analysis yielded a diagnostic yield of 38% for genome-wide sequencing, compared with the diagnostic yield of 21% for standard genetic testing. The risk ratio (RR) is thus in favor of genome-wide sequencing (RR 1.76 [95% CI 1.20–2.58]), with an even larger yield among studies that used GS (43%) than ES (34%). Of note, parents placed strong value on negative test results, emphasizing that diagnostic yield is not a good proxy for parental perceived utility.76 Nondiagnostic results can still be useful to help exclude (or make less likely) genetic conditions that may have prognostic implications (e.g., increased risk of seizures, cancer, or vision loss as examples) or other clinical management (e.g., contraindication to anesthesia/surgery).
Undesirable effects
Potential harms considered in the SER included insurance discrimination; a negative impact on family dynamics or communication; financial burden of the costs associated with additional testing, surveillance, medication, or dietary modifications stemming from the results of ES/GS; general negative psychosocial impact to the patient or their family; and reduction or loss of privacy. However, only five studies in the SER described the harms associated with ES and/or GS (three with n ≥ 20 patients and two studies with n < 20). From a case series of 155 patients, Baldridge et al. reported two cases of misattributed paternity that required ethics consultation and altered strategies for pretest counseling.20 Misattributed paternity was identified in a single case by van Diemen and colleagues requiring the disclosure of misattributed paternity to the family to confirm the diagnosis.23 They reported a case in which following ES, parents declined a potentially therapeutic hematopoietic stem cell transplant for economic reasons.77 Outside of those noted above, the findings across multiple clinical settings suggest no clinically significant psychological harms from the return of ES/GS results. Some populations may experience low levels of test-related distress or greater positive psychological effects.78 Proper consenting process and genetic counseling are important factors that can help mitigate the known associated risks.
Certainty of evidence
While it is clear that there are patients for whom receiving a diagnosis offers direct clinical utility benefits and that patients/families value both the certainty of receiving (or not receiving) a diagnosis, the quality of evidence was formally rated very low because of the paucity of randomized controlled trials and reliance on observational studies and case series that may have higher risk of bias. The quality of evidence evaluating comparative diagnostic yield is moderate. Since the time that the SER and HTA were published, there has been one randomized controlled trial that looked at clinical utility: the second Newborn Sequencing in Genomic Medicine and Public Health (NSIGHT2).35 Physicians in the NSIGHT2 trial reported that rapid genome sequencing (rGS) changed clinical management in 57 (28%) infants, particularly in those receiving ultrarapid genome sequencing (urGS) (p < 0.0001) and positive tests (p < 0.00001). Outcomes of 32 (15%) infants were perceived to be changed by rGS. Positive tests changed outcomes more frequently than negative tests (p < 0.00001). In logistic regression models, the likelihood that rGS was perceived as useful increased 6.7-fold when associated with changes in management (95% CI 1.8–43.3). Changes in management were 10.1-fold more likely when results were positive (95% CI 4.7–22.4) and turnaround time was shorter (odds ratio 0.92, 95% CI 0.85–0.99). rGS seldom led to clinician perceived confusion or distress among families (6 of 207 [3%]).73 This study included some of the population of interest (CA and DD/ID) but was not specific to this population.
Values
Patient preferences and values, obtained through interviews and a review of the qualitative and quantitative evidence, point to consistent motivations and benefits to obtaining a diagnosis for unexplained DD or CA through genome-wide sequencing. Patients and families also greatly value the support and the information provided through genetic counseling when considering genome-wide sequencing and learning of a diagnosis.9
The NSIGHT2 study reported that the large majority of parents felt that first-tier, rapid, diagnostic GS was beneficial for infants lacking etiologic diagnoses in intensive care units (ICUs). Most parents in this study perceived being adequately informed to consent, understood their child’s results, and denied regret or harm from undergoing sequencing.79
Lewis et al. interviewed parents of children with rare diseases participating in the 10,000 Genomes project. Overall, parents were positive about completing the testing for diagnostic purposes. There were more concerns or misunderstandings regarding secondary findings.80
Interviews with 11 parents from the Rapid Paediatric Sequencing (RaPS) study held largely positive views about rGS. They described the clinical and emotional benefits from the opportunity to obtain a rapid diagnosis. Of note, parental stress surrounding their child’s illness complicates their decision making and not unexpectedly their concerns are heightened when offered rGS and waiting for results.81
Balance of effects
In patients with CA/ID/DD, the intervention of ES or GS compared with no ES or GS strongly favors the intervention. There appear to be relatively few harms associated with ES/GS and several valuable benefits.
Resources required
Although studies were published in different years and countries, cost estimates for ES were similar over time and across regions.9,82 It should be noted that the studies did not include the cost of clinic visits, genetic counseling time, ancillary testing, caregiver time, and transportation costs, which impact overall cost-effectiveness in a health-care system. At the same time, direct cost of ES/GS is decreasing over time and cost to the family will vary with insurance coverage and geography.
Cost-effectiveness
The HTA economic model showed that, overall, ES after standard testing increased the diagnostic yield at an additional cost compared to standard testing alone.9,82 However, using ES as a first- or second-tier test (e.g., after CMA or targeted testing) yielded more diagnoses at a lower cost than using ES only after extensive standard testing (e.g., large sequencing panels and/or multiple testing approaches) or using standard testing alone. With the anticipated further declines in cost, early use of genome-wide sequencing should continue to enable more timely diagnosis for patients with unexplained DD or multiple CAs.
Equity
In 2016 the Global Burden of Disease Study reported the total number of children under age 5 years with six DDs was 52.9 million, with 50.2 million (94.9%) in low and middle income countries, and 2.7 million (5.1%) in high income countries.83 There does not appear to be any empirical evidence specifically regarding equity for ES/GS. A PubMed search returned 0 applicable results for “health equity” and the available MESH terms “whole exome sequencing” or “whole genome sequencing.” However, it is well-established that minority populations are historically underrepresented in genomic studies.84 Patients with health insurance and from higher socioeconomic backgrounds are more likely to have access and pursue genetic services. On the other hand, given socioeconomic status–based inequities in access to care, clinical experience suggests that if a diagnosis can be made in fewer visits, increased use of ES/GS should increase equity for patients with genetic disorders.
Acceptability
Based on the studies listed in the SER, HTA, and other references in these assessments, key stakeholders including health-care providers, patients/families, and laboratories find ES/GS is acceptable.
Feasibility
The number of published studies in the SER, HTA, and other references in these assessments have shown the technical and logistical feasibility of ES and GS.
Subgroup considerations
Consistent with existing guidelines/recommendations/position statements, patients with clinical presentations highly suggestive of a specific genetic diagnosis should undergo targeted testing first. This may include patients with suspicion of a chromosomal disorder, known family history of a disorder, or strong clinical suspicion of a diagnosis in which sequencing may not be diagnostic, such as Prader–Willi/Angelman related methylation abnormality or fragile X syndrome.
The use of rapid and ultrarapid (currently defined as 6–15 days and 1–3 days, respectively) ES/GS is also an evolving area as there may be some situations where early diagnosis can make a significant and immediate difference. Cost and quality of interpretation are significant factors in the risk/benefit analysis, but rapid testing should also be made available if clinical indications regarding utility are met. In patients with clinical symptoms requiring acute management, rapid turnaround testing might obviate the need for more extensive and expensive diagnostic workups, make a patient eligible for a targeted therapy, or may allow clinicians to avoid subjecting the patient to ineffective diagnostic or therapeutic interventions with potential for unwanted side effects. Arriving at a diagnosis may also clarify prognosis and natural history in such a manner that allows parents and medical teams to make informed decisions about the nature and goals of care.
Isolated autism without ID or congenital malformation is formally out of scope for this recommendation but evaluation of exome/genome studies is ongoing.
Implementation considerations
The value of genetic counseling in ES/GS is well-established.85,86 Creating reasonable expectations, establishing an understanding of the value and limitations of testing, creating awareness of the potential harms, and allowing the family to make informed choices is a mainstay of informed consent for ES/GS. These visits should also be commensurate with the time spent as part of the clinical process including reimbursement for this type of counseling. Post-test counseling extends this benefit once the results are available regardless of the diagnostic yield. Elements of counseling should include a three-generation family pedigree; discussion of pathogenic/likely pathogenic results, benign results, and variants of uncertain significance; detection of misattributed paternity or consanguinity, and secondary findings unrelated to the reason for testing.
The ACMG Secondary Findings v3.0 is the recently released minimum set of genes recommended for evaluation in a diagnostic exome or genome.7,8 Carrier status is reported by some labs as a secondary finding. All secondary findings should be available but are optional. Because of testing of family members as comparators, there is the potential to identify risk for a genetic disease in an unaffected parent. Limits of testing should be discussed, including limited disease–gene known associations. Post-test counseling should include the opportunity for reanalysis and reclassification of variants that may lead to amended interpretation and issuing a new report.
Monitoring and evaluation
Re-evaluation of the evidence base for ES and GS should be done at regular intervals given rapid technological advancements leading to improved diagnostic yield, variant interpretation, and reduction in cost.87 The exact interval of this should consider the resources required for another systematic review including time required and cost. Living systematic reviews, where new evidence is added to the existing literature base as studies are published, may allow ACMG to update this guideline more nimbly in the future. It would be reasonable to expect that yield for ES/GS would increase based on the current progress. There are preliminary clinical data to demonstrate similar performance of GS to ES+CMA for diagnostic yield and potential for lower cost for GS, which would depend on sequential or concurrent ES+CMA.88,89,90
Measuring change to practice will be valuable but since testing is both commercial and health system based, comprehensive test utilization measures may not easily be achievable or accessible. Surveying clinical geneticists and labs as to ability to apply guidelines may provide a surrogate marker but would be still imprecise. Developing a monitoring program for health disparities should also be addressed. Cost-effectiveness should be reassessed as well using systematic review as the literature increases but interim findings are encouraging.91 Workforce considerations will change including ordering providers, need of clinical geneticists/genetic counselors, and lab availability.
Research priorities
There are some limitations to this analysis that could be addressed with future research. While there is growing evidence of the value of reanalysis of previous ES/GS, the timing and optimal strategy are not clear at this time. Improvements in analytic tools, advancements in literature, and updates to reference databases are but some of the considerations. Pairing genomic analysis with other -omic approaches (e.g., transcriptome, methylome) may improve interpretability, yield, and actionability. There is also mounting research evidence that in selected cases, paired transcriptome sequencing with ES/GS has the potential to boost diagnostic yields by improving recovery/interpretability of disease-relevant variants that have impact at the messenger RNA (mRNA) or pre-mRNA level.92,93 Similarly, advances in algorithms that use GS data to identify copy-number variants and structural and other complex variants will also improve diagnostic yield. Clinical management will continue to improve as natural history is better understood to allow for better screening and anticipation of complications of rare disorders. But more substantially, advancements in treatments targeting specific genetic variants (e.g., elexacaftor–tezacaftor–ivacaftor for cystic fibrosis), general classes of variants (readthrough therapies, etc.), or gene therapy modalities will only enhance clinical utility.
Evaluation of ES/GS beyond CA and ID including for isolated autism, cardiomyopathies, muscular dystrophies, neuropathies, ataxias, epilepsies, and inherited cancers would be expected to demonstrate similar clinical utility. Additionally, somatic variants that may not be revealed by germline testing but that may be the cause of disease could be considered for future third-tier evaluation. Capturing these management impacts via the implementation of learning health-care system models is vital, as the separation of research and clinical testing is in constant flux.
The systematic review process identified important gaps in the literature regarding family-focused outcomes and lab considerations such as standards of practice and the importance of sharing variant interpretation in open databases to improve diagnostic yield and reduce VUS. Given the fractured nature of health-care delivery in the United States, continued provider education will be an important component to adoption of ES/GS as a first- or second-line test for patients with CA/DD/ID. Health-care equity is lacking in the field overall. In the implementation of this recommendation, care should be taken to avoid exacerbating existing health disparities based on unequal access to care as well as inadequate data to support variant interpretation in populations underrepresented by current sequencing efforts.
The Genetic Information Nondiscrimination Act (GINA) provided legal protections for employee/health insurance protection based on early concerns for expansion of genetic testing and reduces possible harms with testing. Expanded application for GINA for long-term care and other forms of discrimination should be an important consideration.
Conclusions
In summary, based on the SER there is a strong recommendation based on the available evidence to support the use of ES/GS as either a first- (or second-) line test in patients with CA/DD/ID. ES/GS demonstrates clinical utility for the patients and their families with limited evidence for negative outcomes and the ever-increasing emerging evidence of therapeutic benefit.
References
Mokdad, A. H. et al. Global burden of diseases, injuries, and risk factors for young people’s health during 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 387, 2383–2401 (2016).
Dolk, H. EUROCAT: 25 years of European surveillance of congenital anomalies. Arch. Dis. Child. Fetal. Neonatal. Ed. 90, F355–358 (2005).
Malinowski, J. et al. Systematic evidence-based review: outcomes from exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability. Genet. Med. 22, 986–1004 (2020).
Institute of Medicine (US) Committee on Standards for Developing Trustworthy Clinical Practice Guidelines. Clinical Practice Guidelines We Can Trust. (The National Academies Press, Washington, DC, 2011).
Manning, M. & Hudgins, L. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet. Med. 12, 742–745 (2010).
Delanne, J. et al. Secondary findings from whole-exome/genome sequencing evaluating stakeholder perspectives. A review of the literature. Eur. J. Med. Genet. 62, 103529 (2019).
Miller, D. T. et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2021 update: a policy statement of the American College of Medical Genetics and Genomics. Genet. Med. https://doi.org/10.1038/s41436-021-01171-4 (2021).
Miller, D. T. et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. https://doi.org/10.1038/s41436-021-01172-3 (2021).
Ontario Health. Genome-wide sequencing for unexplained developmental disabilities or multiple congenital anomalies: a health technology assessment. Ont. Health Technol. Assess. Ser. 20, 1–178 (2020).
Schunemann, H. J. et al. GRADE Guidelines: 16. GRADE evidence to decision frameworks for tests in clinical practice and public health. J. Clin. Epidemiol. 76, 89–98 (2016).
Farnaes, L. et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom. Med. 3, 10 (2018).
Perucca, P. et al. Real-world utility of whole exome sequencing with targeted gene analysis for focal epilepsy. Epilepsy Res. 131, 1–8 (2017).
Petrikin, J. E., Willig, L. K., Smith, L. D. & Kingsmore, S. F. Rapid whole genome sequencing and precision neonatology. Semin. Perinatol. 39, 623–631 (2015).
Sawyer, S. L. et al. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin. Genet. 89, 275–284 (2016).
Scocchia, A. et al. Clinical whole genome sequencing as a first-tier test at a resource-limited dysmorphology clinic in Mexico. NPJ Genom. Med. 4, 5 (2019).
Tarailo-Graovac, M. et al. Exome sequencing and the management of neurometabolic disorders. N. Engl. J. Med. 374, 2246–2255 (2016).
Zhu, X. et al. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet. Med. 17, 774–781 (2015).
Kuperberg, M. et al. Utility of whole exome sequencing for genetic diagnosis of previously undiagnosed pediatric neurology patients. J. Child Neurol. 31, 1534–1539 (2016).
Miller, K. A. et al. Diagnostic value of exome and whole genome sequencing in craniosynostosis. J. Med. Genet. 54, 260–268 (2017).
Baldridge, D. et al. The Exome Clinic and the role of medical genetics expertise in the interpretation of exome sequencing results. Genet. Med. 19, 1040–1048 (2017).
Srivastava, S. et al. Clinical whole exome sequencing in child neurology practice. Ann. Neurol. 76, 473–483 (2014).
Bourchany, A. et al. Reducing diagnostic turnaround times of exome sequencing for families requiring timely diagnoses. Eur. J. Med. Genet. 60, 595–604 (2017).
van Diemen, C. C. et al. Rapid targeted genomics in critically ill newborns. Pediatrics. 140, e20162854 (2017).
Iglesias, A. et al. The usefulness of whole-exome sequencing in routine clinical practice. Genet. Med. 16, 922–931 (2014).
Nolan, D. & Carlson, M. Whole exome sequencing in pediatric neurology patients: clinical implications and estimated cost analysis. J. Child Neurol. 31, 887–894 (2016).
Valencia, C. A. et al. Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: a Pediatric Center’s experience. Front. Pediatr. 3, 67 (2015).
Meng, L. et al. Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. 171, e173438 (2017).
Stark, Z. et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet. Med. 18, 1090–1096 (2016).
Cordoba, M. et al. Whole exome sequencing in neurogenetic odysseys: an effective, cost- and time-saving diagnostic approach. PLoS One. 13, e0191228 (2018).
French, C. E. et al. Whole genome sequencing reveals that genetic conditions are frequent in intensively ill children. Intensive Care Med. 45, 627–636 (2019).
Soden, S. E. et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci. Transl. Med. 6, 265ra168 (2014).
Stark, Z. et al. Meeting the challenges of implementing rapid genomic testing in acute pediatric care. Genet. Med. 20, 1554–1563 (2018).
Thevenon, J. et al. Diagnostic odyssey in severe neurodevelopmental disorders: toward clinical whole-exome sequencing as a first-line diagnostic test. Clin. Genet. 89, 700–707 (2016).
Willig, L. K. et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir. Med. 3, 377–387 (2015).
Petrikin, J. E. et al. The NSIGHT1-randomized controlled trial: rapid whole-genome sequencing for accelerated etiologic diagnosis in critically ill infants. NPJ Genom. Med. 3, 6 (2018).
Bick, D. et al. Successful application of whole genome sequencing in a Medical Genetics Clinic. J. Pediatr. Genet. 6, 61–76 (2017).
Scheuner, M. T. et al. Stakeholders’ views on the value of outcomes from clinical genetic and genomic interventions. Genet. Med. 21, 1371–1380 (2019).
Anazi, S. et al. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol. Psychiatry. 22, 615–624 (2017).
Bekheirnia, M. R. et al. Whole-exome sequencing in the molecular diagnosis of individuals with congenital anomalies of the kidney and urinary tract and identification of a new causative gene. Genet. Med. 19, 412–420 (2017).
Brett, G. R. et al. Parental experiences of ultrarapid genomic testing for their critically unwell infants and children. Genet. Med. 22, 1976–1985 (2020).
Ceyhan-Birsoy, O. et al. Interpretation of genomic sequencing results in healthy and ill newborns: results from the BabySeq Project. Am. J. Hum. Genet. 104, 76–93 (2019).
Cheng, S. S. W. et al. Experience of chromosomal microarray applied in prenatal and postnatal settings in Hong Kong. Am. J. Med. Genet. C Semin. Med. Genet. 181, 196–207 (2019).
Clark, M. M. et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom. Med. 3, 16 (2018).
Dixon-Salazar, T. J. et al. Exome sequencing can improve diagnosis and alter patient management. Sci. Transl. Med. 4, 138ra178 (2012).
Evers, C. et al. Impact of clinical exomes in neurodevelopmental and neurometabolic disorders. Mol. Genet. Metab. 121, 297–307 (2017).
Fan, L. L. et al. Whole exome sequencing identifies a novel mutation (c.333+2T>C) of TNNI3K in a Chinese family with dilated cardiomyopathy and cardiac conduction disease. Gene. 648, 63–67 (2018).
Hochstenbach, R. et al. Array analysis and karyotyping: workflow consequences based on a retrospective study of 36,325 patients with idiopathic developmental delay in the Netherlands. Eur. J. Med. Genet. 52, 161–169 (2009).
Husereau, D. et al. Consolidated Health Economic Evaluation Reporting Standards (CHEERS) statement. BMJ. 346, f1049 (2013).
Jain, P., Sharma, S. & Tripathi, M. Diagnosis and management of epileptic encephalopathies in children. Epilepsy Res. Treat. 2013, 501981 (2013).
Jang, W. et al. Chromosomal microarray analysis as a first-tier clinical diagnostic test in patients with developmental delay/intellectual disability, autism spectrum disorders, and multiple congenital anomalies: a prospective multicenter study in Korea. Ann. Lab. Med. 39, 299–310 (2019).
Kaye, A. J., Rand, E. B., Munoz, P. S., Spinner, N. B., Flake, A. W. & Kamath, B. M. Effect of Kasai procedure on hepatic outcome in Alagille syndrome. J. Pediatr. Gastroenterol. Nutr. 51, 319–321 (2010).
Kingsmore, S. F. et al. A randomized, controlled trial of the analytic and diagnostic performance of singleton and trio, rapid genome and exome sequencing in ill infants. Am. J. Hum. Genet. 105, 719–733 (2019).
Kohler, J. N. et al. Defining personal utility in genomics: a Delphi study. Clin. Genet. 92, 290–297 (2017).
Malek, J. et al. Responsibility, culpability, and parental views on genomic testing for seriously ill children. Genet. Med. 21, 2791–2797 (2019).
Mollison, L., O’Daniel, J. M., Henderson, G. E., Berg, J. S. & Skinner, D. Parents’ perceptions of personal utility of exome sequencing results. Genet. Med. 22, 752–757 (2020).
Nair, P. et al. Contribution of next generation sequencing in pediatric practice in Lebanon. A study on 213 cases. Mol. Genet. Genomic Med. 6, 1041–1052 (2018).
Nambot, S. et al. Clinical whole-exome sequencing for the diagnosis of rare disorders with congenital anomalies and/or intellectual disability: substantial interest of prospective annual reanalysis. Genet. Med. 20, 645–654 (2018).
Nemirovsky, S. I. et al. Whole genome sequencing reveals a de novo SHANK3 mutation in familial autism spectrum disorder. PLoS One. 10, e0116358 (2015).
Palmer, E. E. et al. Integrating exome sequencing into a diagnostic pathway for epileptic encephalopathy: evidence of clinical utility and cost effectiveness. Mol. Genet. Genomic Med. 6, 186–199 (2018).
Pereira, S. et al. Perceived benefits, risks and utility of newborn genomic sequencing in the BabySeq project. Pediatrics. 143, S6–S13 (2019).
Powis, Z. et al. Exome sequencing in neonates: diagnostic rates, characteristics, and time to diagnosis. Genet. Med. 20, 1468–1471 (2018).
Rosell, A. M. et al. Not the end of the odyssey: parental perceptions of whole exome sequencing (WES) in pediatric undiagnosed disorders. J. Genet. Couns. 25, 1019–1031 (2016).
Shamseer, L. et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015: elaboration and explanation. BMJ. 350, g7647 (2015).
Smith, H. S. et al. Clinical application of genome and exome sequencing as a diagnostic tool for pediatric patients: a scoping review of the literature. Genet. Med. 21, 3–16 (2019).
Tammimies, K. et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA. 314, 895–903 (2015).
Tan, T. Y. et al. Diagnostic impact and cost-effectiveness of whole-exome sequencing for ambulant children with suspected monogenic conditions. JAMA Pediatr. 171, 855–862 (2017).
Thiffault, I. et al. Clinical genome sequencing in an unbiased pediatric cohort. Genet. Med. 21, 303–310 (2019).
Todd, E. J. et al. Next generation sequencing in a large cohort of patients presenting with neuromuscular disease before or at birth. Orphanet. J. Rare. Dis. 10, 148 (2015).
Vissers, L. E. L. M. et al. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet. Med. 19, 1055–1063 (2017).
Wang, H. et al. Clinical utility of 24-h rapid trio-exome sequencing for critically ill infants. NPJ Genom. Med. 5, 20 (2020).
Guyatt, G. et al. GRADE guidelines: 1. Introduction-GRADE evidence profiles and summary of findings tables. J. Clin. Epidemiol. 64, 383–394 (2011).
Guyatt, G. H. et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ. 336, 924–926 (2008).
Zhang, Y., Akl, E. A. & Schunemann, H. J. Using systematic reviews in guideline development: the GRADE approach. Res. Synth. Methods. 10, 312–329 (2019).
Schwarzer, G., Chemaitelly, H., Abu-Raddad, L. J. & Rucker, G. Seriously misleading results using inverse of Freeman-Tukey double arcsine transformation in meta-analysis of single proportions. Res. Synth. Methods. 10, 476–483 (2019).
Warton, D. I. & Hui, F. K. C. The arcsine is asinine: the analysis of proportions in ecology. Ecology. 92, 3–10 (2011).
Cakici, J. A. et al. A prospective study of parental perceptions of rapid whole-genome and -exome sequencing among seriously ill infants. Am. J. Hum. Genet. 107, 953–962 (2020).
He, X., Zou, R., Zhang, B., You, Y., Yang, Y. & Tian, X. Whole Wiskott-Aldrich syndrome protein gene deletion identified by high throughput sequencing. Mol. Med. Rep. 16, 6526–6531 (2017).
Robinson, J. O. et al. Psychological outcomes related to exome and genome sequencing result disclosure: a meta-analysis of seven Clinical Sequencing Exploratory Research (CSER) Consortium studies. Genet. Med. 21, 2781–2790 (2019).
Dimmock, D. P. et al. An RCT of rapid genomic sequencing among seriously ill infants results in high clinical utility, changes in management, and low perceived harm. Am. J. Hum. Genet. 107, 942–952 (2020).
Lewis, C. et al. Parents’ motivations, concerns and understanding of genome sequencing: a qualitative interview study. Eur. J. Hum. Genet. 28, 874–884 (2020).
Hill, M., Hammond, J., Lewis, C., Mellis, R., Clement, E. & Chitty, L. S. Delivering genome sequencing for rapid genetic diagnosis in critically ill children: parent and professional views, experiences and challenges. Eur. J. Hum. Genet. 28, 1529–1540 (2020).
Schwarze, K., Buchanan, J., Taylor, J. C. & Wordsworth, S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet. Med. 20, 1122–1130 (2018).
Global Research on Developmental Disabilities Collaborators. Developmental disabilities among children younger than 5 years in 195 countries and territories, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Glob. Health. 6, e1100–e1121 (2018).
Popejoy, A. B. & Fullerton, S. M. Genomics is failing on diversity. Nature. 538, 161–164 (2016).
Macnamara, E. F. et al. Cases from the Undiagnosed Diseases Network: the continued value of counseling skills in a new genomic era. J. Genet. Couns. 28, 194–201 (2019).
Madlensky, L., Trepanier, A. M., Cragun, D., Lerner, B., Shannon, K. M. & Zierhut, H. A rapid systematic review of outcomes studies in genetic counseling. J. Genet. Couns. 26, 361–378 (2017).
Deignan, J. L. et al. Points to consider in the reevaluation and reanalysis of genomic test results: a statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 21, 1267–1270 (2019).
Lindstrand, A. et al. From cytogenetics to cytogenomics: whole-genome sequencing as a first-line test comprehensively captures the diverse spectrum of disease-causing genetic variation underlying intellectual disability. Genome Med. 11, 68 (2019).
Lionel, A. C. et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet. Med. 20, 435–443 (2018).
Stranneheim, H. et al. Integration of whole genome sequencing into a healthcare setting: high diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Med. 13, 40 (2021).
Carey, A. S. et al. Rapid exome sequencing in PICU patients with new-onset metabolic or neurological disorders. Pediatr. Res. 88, 761–768 (2020).
Cummings, B. B. et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci. Transl. Med. 9, eaal5209 (2017).
Wong, M. et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat. Med. 26, 1742–1753 (2020).
Author information
Authors and Affiliations
Consortia
Ethics declarations
Competing interests
H.M.K. is a director of a clinical laboratory that performs a breadth of genetic and genomic analyses on a fee-for service basis. K.M., L.A.D., S.B., L.J.M., T.W.Y., and F.M.H. clinically see patients with rare disorders. D.M. is the head of a foundation for a rare disorder and a parent of two children with a rare disorder. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Disclaimer
The ACMG has recruited expert panels, chosen for their scientific and clinical expertise, to develop evidence-based guidelines (EBG) for clinical practice. An EBG focuses on a specific scientific question and then describes recommendations intended to optimize patient care that are informed by a systematic review of evidence and an assessment of the benefits and harms of alternative care options. ACMG EBGs are provided primarily as an educational resource for medical geneticists and other clinicians to help them provide quality medical services. They should not be considered inclusive of all relevant information on the topic reviewed.
Reliance on this EBG is completely voluntary and does not necessarily ensure a successful medical outcome. In determining the propriety of any specific procedure or test, the clinician should consider the best available evidence, and apply his or her own professional judgment, taking into account the needs, preferences and specific clinical circumstances presented by the individual patient. Clinicians are encouraged to document the reasons for the use of a particular procedure or test, whether or not it is in conformance with this EBG. Clinicians are also advised to take notice of the date this EBG was published, and to consider other medical and scientific information that becomes available after that date.
*The Board of Directors of the American College of Medical Genetics and Genomics approved this evidence-based guideline on 24 May 2021.
Rights and permissions
About this article

Cite this article
Manickam, K., McClain, M.R., Demmer, L.A. et al. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med 23, 2029–2037 (2021). https://doi.org/10.1038/s41436-021-01242-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-021-01242-6
This article is cited by
-
Genetic and phenotypic analysis of 225 Chinese children with developmental delay and/or intellectual disability using whole-exome sequencing
BMC Genomics (2024)
-
Exome sequencing improves the molecular diagnostics of paediatric unexplained neurodevelopmental disorders
Orphanet Journal of Rare Diseases (2024)
-
Optimizing genetic testing strategies for congenital anomalies in Iran
European Journal of Human Genetics (2024)
-
Advancing access to genome sequencing for rare genetic disorders: recent progress and call to action
npj Genomic Medicine (2024)
-
Evidence review and considerations for use of first line genome sequencing to diagnose rare genetic disorders
npj Genomic Medicine (2024)
