
Abstract
Purpose
Within this study, we aimed to discover novel gene–disease associations in patients with no genetic diagnosis after exome/genome sequencing (ES/GS).
Methods
We followed two approaches: (1) a patient-centered approach, which after routine diagnostic analysis systematically interrogates variants in genes not yet associated to human diseases; and (2) a gene variant centered approach. For the latter, we focused on de novo variants in patients that presented with neurodevelopmental delay (NDD) and/or intellectual disability (ID), which are the most common reasons for genetic testing referrals. Gene–disease association was assessed using our data repository that combines ES/GS data and Human Phenotype Ontology terms from over 33,000 patients.
Results
We propose six novel gene–disease associations based on 38 patients with variants in the BLOC1S1, IPO8, MMP15, PLK1, RAP1GDS1, and ZNF699 genes. Furthermore, our results support causality of 31 additional candidate genes that had little published evidence and no registered OMIM phenotype (56 patients). The phenotypes included syndromic/nonsyndromic NDD/ID, oral–facial–digital syndrome, cardiomyopathies, malformation syndrome, short stature, skeletal dysplasia, and ciliary dyskinesia.
Conclusion
Our results demonstrate the value of data repositories which combine clinical and genetic data for discovering and confirming gene–disease associations. Genetic laboratories should be encouraged to pursue such analyses for the benefit of undiagnosed patients and their families.
Similar content being viewed by others


INTRODUCTION
More than half of patients with genetic diseases remain undiagnosed, even after conducting genome-wide diagnostic approaches, such as exome and genome sequencing.1,2 Despite recent technological advances, the challenge of variant interpretation remains, in part due to the missing gene–phenotype link.3
The methods applied for the identification of causal gene defects for monogenic diseases have changed drastically in the last 10 years. Genome-wide scans using polymorphic microsatellite markers or single-nucleotide variants followed by linkage analysis were the predominant genetic mapping approach used in the past.4 This changed dramatically after the implementation and routine application of exome/genome sequencing in genetic research. Currently, most family-based approaches for disease gene identification rely on the analysis of exome or genome data. Study designs vary from including single unrelated individuals with a similar phenotype to typical family-based studies with the inclusion of several affected and unaffected relatives to focus on regions of homozygosity or using the de novo approach.5 Furthermore, phenocentric (focused on specific patients and phenotypes) and genocentric (focused on database analyses and algorithms) approaches have been described.6,7 Identification of candidate genes/variants associated with disease is usually followed by replication in other unrelated, similarly affected patients and/or functional studies to validate variants’ pathogenicity.5
The unambiguous assignment of disease causality is often difficult to achieve, and, in many cases, the initially collected evidence is insufficient to prove causality. The rarity, severity, and clinical heterogeneity of many genetic disorders complicates the process of finding additional patients. Furthermore, the lack of knowledge on the gene/protein function challenges the final assignment of gene causality. Thus, the gene candidacy remains inconclusive and is considered as a research gene.
Within this study, we analyzed exome/genome data together with the respective clinical phenotypes of the patients using Human Phenotype Ontology (HPO) to identify novel gene–disease associations and to validate previously reported candidate genes. We present six novel gene–disease associations and the confirmation of 31 additional candidate genes. The outcome has substantial implications for the diagnosis and counseling of the patients and their families.
MATERIALS AND METHODS
Patients
Written informed consent included several sections: consent for genetic testing related to the disease(s) of the patient, and consent for research (related to the main concern, but implicating genes not yet associated to human diseases). Additionally, the consent declaration included information regarding storage of the data and further processing for research purposes. Written informed consent was given by patients, parents, or referring physicians. Consent for scientific publication of patient photographs was obtained as well. Data regarding country of origin, family history, consanguinity, clinical phenotype, and previous genetic testing were extracted from our database.
Exome and genome sequencing (ES/GS)
DNA was extracted from EDTA blood or from dried blood spots on filter cards (CentoCard®) using standard, spin column-based methods.
ES was performed as previously described.2 In short, the Nextera Rapid Capture Exome Kit (Illumina, San Diego, CA), the SureSelect Human All Exon kit (Agilent, Santa Clara, CA) or the Twist Human Core Exome was used for enrichment, and a Nextseq500, HiSeq4000, or Novoseq 6000 (Illumina) instrument was used for the actual sequencing, with the average coverage targeted to at least 100× or at least 98% of the target DNA covered 20×. When carrying out GS, genomic DNA was fragmented by sonication, and Illumina adapters were ligated to generated fragments for subsequent sequencing on the HiSeqX platform (Illumina) to yield an average coverage depth of at least 30×. Data analysis, including base calling, de-multiplexing, alignment to the hg19 human reference genome (Genome Reference Consortium GRCh37), and variant calling, was performed using the HiSeq Analysis Software v2.0 pipelines (Illumina, Inc., San Diego, CA), as previously described8 (Supplementary information).
Variants with suboptimal quality were confirmed via Sanger sequencing according to our established criteria9 or quantitative polymerase chain reaction (qPCR), multiplex ligation-dependent probe amplification (MLPA), or chromosomal microarray (CMA) for copy-number variations (CNVs). An extended Methods section can be found in the Supplementary Information.
Variant evaluation and classification
The clinical information was translated into HPO terms, registered in our data repository, and applied for each individual analysis during variant filtration and prioritization as previously described.2,10 Variant nomenclature followed standard Human Genome Variation Society (HGVS) recommendations.11 Variants in established diagnostic genes were classified according to the published guidelines of the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) as pathogenic (P), likely pathogenic (LP), and variant of unknown significance (VUS).12
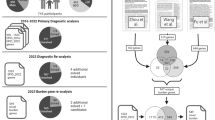
For patients with no relevant variant(s) identified during the diagnostic process, a second analysis was conducted with the aim of identifying variants in genes not yet associated to any human phenotype. The results were reported to the referring physician as research findings in a dedicated section of the genetic report. The workflow is summarized in Fig. 1a.

a Patient-centered approach to systematically interrogate variants in genes not yet associated to human diseases in patients with no genetic diagnosis after exome/genome sequencing (ES/GS). b Gene variant centered approach to analyze de novo variants in cases with ES/GS performed and no genetic diagnosis. DP depth of reads, pLI probability of loss-of-function intolerance.
Analysis of own data repository
Patients’ reports containing research findings were retrieved from the database (2016–2019). Reported variants were reassessed taking into consideration current knowledge on the gene function and compatibility with the patient phenotype (e.g., based on animal models). Only cases with negative or inconclusive (VUS) diagnostic findings were included (Fig. 1a). As a second step, the data repository was queried for other rare variants in the respective candidate gene and the overlapping clinical features of the individuals.
Our data repository (CentoMD®)13 contains ES/GS data from 55,782 individuals (50,023 ES/5,759 GS), of whom 33,280 individuals (29,842 ES/3,438 GS) have clinical descriptions that include at least one HPO term. Neurodevelopmental delay (NDD) or intellectual disability (ID) are among the most frequent reasons for genetic consultation and testing. Thus, a second, gene-centered approach was applied to identify de novo variants in patients with NDD/ID. Variants that are rare in external databases (ExAc ≤0.0001 or gnomAD ≤0.0001) and have a high or moderate predicted impact on protein structure or function (missense, affecting splicing sites, nonsense, frameshift, indels) and high CADD raw score (above 4) were prioritized. Only variants with satisfactory quality scores were considered (read depth ≥20, frequency ≥20 and quality score ≥220).9 In addition, variants mapping to 3,230 genes with high probability of loss-of-function intolerance (pLI) scores ExAC calculations of pLI ≥0.90) were prioritized. Genes with an associated clinical phenotype in OMIM or ClinVar were excluded from this analysis. Finally, only index cases with parental samples available who had no established genetic diagnosis during former ES/GS evaluations were included. Figure 1b summarizes the filtering strategy.
RESULTS
We applied two different strategies to identify novel gene–disease associations. For the first approach (patient-centered), we extended the ES/GS evaluation to genes with no known disease association, according to the OMIM database. A summary of the implemented workflow is shown in Fig. 1a. In using this strategy, we identified 191 candidate genes in patients with a wide range of clinical phenotypes. Furthermore, we used a second approach (gene-centered) oriented toward identifying unreported, de novo variants in patients with NDD/ID. We focused on NND/ID as these are among the main reasons for genetic testing referrals. The main parameters applied are summarized in Fig. 1b. We identified 287 candidate genes using this approach.
Then, we reviewed the evidence supporting variant/gene pathogenicity and individual patient data. We took into consideration the OMIM database, PubMed, Uniprot, and the Human Protein Atlas. With this evaluation, we detected genes that were already recognized by us as candidates and for which independent publications were ongoing, for example ADAMTS1914 and EMC10.15 Additional genes had recently been published as causal for genetic disorders, such as FBXW11,16 GRIA2,17 PPP1R21,18 and TAOK1, which was recently published by us.19 Other genes such as TANC220 and NEK1021 were published as causative in the months following our initial analysis and during the preparation of this paper. These examples can be considered as a proof of principle, confirming the effectiveness of the applied approaches.
For the identification of novel gene–disease associations, we focused on genes with more than one hit and no previous association to a human disease. We selected genes with variants in at least two unrelated cases and published genetic or functional evidence indicating a role in disease, or with at least three unrelated patients if there was limited available evidence on gene function. This analysis enabled the identification of novel gene–disease associations based on 38 patients with variants in six genes: BLOC1S1, IPO8, MMP15, PLK1, RAP1GDS1, and ZNF699 (Table 1).
The related disorders are described as follows: three different neurodevelopmental disorders with features of (1) severe ID, leukodystrophy, seizures, and visual impairment (BLOC1S1, four patients); (2) NDD, seizures, and microcephaly (PLK1, five patients); and (3) NDD, dysmorphic features, and hypotonia (RAP1GDS1, four patients). We also identified three new syndromic associations: (1) a connective tissue disorder resembling Loeys–Dietz syndrome (LDS) (IPO8, nine patients); (2) Alagille-like syndrome with liver cholestasis and congenital heart defects (MMP15, three patients); and (3) a multiple malformation syndrome (ZNF699, 13 patients). Variant details and patient phenotypes are summarized in Table 1. All cases presented homozygous variants compatible with autosomal recessive disorders. Selected examples are described below.
BLOC1S1
Four patients from three families presented rare homozygous variants in this gene and a similar neurological phenotype. Additional testing in one family confirmed cosegregation of the variant in two siblings (affected sibling—homozygote and unaffected sibling—heterozygote). BLOC1S1 is a component of the ubiquitously expressed BLOC1 multisubunit protein complex, which is required for normal biogenesis of specialized organelles of the endosomal–lysosomal system.22 The gene was originally identified as GCN5L1; it has been shown to play crucial roles in mitochondria, endosomes, lysosomes, and synaptic vesicle precursors.23 Knocking out this gene in mice results in lethality; mice embryos fail to develop beyond ∼E12.5.24 Furthermore, mutant flies lacking the conserved Blos1 subunit displayed eye pigmentation defects, as well as abnormal glutamatergic transmission and behavior.25 BLOC1S3 is another component of the ubiquitously expressed BLOC1 multisubunit protein complex. Biallelic pathogenic variants in BLOC1S3 cause Hermansky–Pudlak syndrome (OMIM 614077), a sort of incomplete oculocutaneous albinism and platelet dysfunction that includes visual defects.
IPO8
Six different homozygous LoF variants were identified in the IPO8 gene in nine unrelated patients (Table 1 and Fig. 2). Phenotypically, the patients presented dysmorphic features, hypotonia, and features reminiscent of a connective tissue disease such as high palate, pectus deformities, hernias, gray-blue sclera, cutis laxa, tortuosity of cerebral arteries, and congenital heart defects. For some cases, clinical suspicion included LDS and Ehlers–Danlos syndrome. The IPO8 gene has not been associated to any human phenotype so far. Interestingly, Imp8 is involved in preferential nuclear importing of Smad1, Smad3, and Smad4. The TGFB pathway and receptor SMADs (SMAD2/3) are central in the pathophysiology of LDS with causative variants detected in the TGFBR2/3,26 TGFB2/3,27,28 SMAD2/3.29,30

Upper panel: photographs illustrating clinical features. Patient 7, with umbilical hernia, brachydactyly of hands, proximal placement of the thumbs, short nails, genus varus, pes planus, brachydactyly of foot, and short toenails. Patient 9 with low-set ears, sparse scalp hair, broad and sparse eyebrows, hypertelorism, long palpebral fissures, and depressed nasal bridge. Patient 13 with frontal bossing, wide, sparse eyebrows, hypertelorism, large palpebral fissures (downslanted), deep philtrum, and thin vermilion of the upper lip. Joint hypermobility (wrist and thumb), as well as long foot, long toes, hindfoot valgus, and pes planus. Lower panel: family trees of patients with IPO8 homozygous LoF variants and clinical abnormalities. Patients presented with a complex phenotype that included abnormalities of the cardiovascular system (congenital heart defects, cardiomyopathy, engorged brain vasculature), the skeletal system (joint hypermobility, pectus deformities, genus valgus/varus, scoliosis), and the skin (cutis laxa). Most patients presented hypotonia, neurodevelopmental delay (NDD), and failure to thrive. Other features included intestinal malrotation, Gastroesophageal reflux (GER), and hydronephrosis.
MMP15
Upon the detection of the homozygous variant NM_002428.3:c.1058delC, p.Pro353fs in a patient with dysmorphic features, complex congenital heart defects (double outlet of the right ventricle, hypoplastic left ventricle, septal defects), and cholestasis, we queried our data repository for additional cases. A sibling was similarly affected and was homozygote for the same variant. An additional unrelated patient was identified with a different variant in the MMP15 gene (Table 1). The patient presented cholestasis, hepatomegaly, high hepatic transaminases, and congenital heart disease. Alagille syndrome and progressive familial intrahepatic cholestasis were the differential diagnoses. MMP15, a member of the matrix metalloproteinases family, is an excellent candidate for this phenotype. In mice, Mmp15 is a direct target of Snail1 during endothelial to mesenchymal transformation and endocardial cushion development.31 A Snail1/Notch1 signaling axis controls embryonic vascular development. Snail1 acts as a VEGF-induced regulator of Notch1 signaling and Dll4 expression.32 In humans, genes from the NOTCH pathway (JAG1 and NOTCH2) are implicated in Alagille syndrome type 1 and 2 (OMIM 118450 and 610205), which has high similarity with the phenotype described here in patients with homozygous variants in MMP15. Interestingly, while these syndromes present with an autosomal dominant mode of inheritance, the patients reported in this study with MMP15 variants show an autosomal recessive disease.
ZNF699
Thirteen patients from 12 families were identified with homozygous loss-of-function (LoF) variants in this gene (Fig. 3). These patients presented with a clear malformation syndrome with coarse facial features and abnormalities of the cardiovascular, gastrointestinal (gastroesophageal reflux, intestinal atresia), genitourinary (renal dysplasia/hypoplasia, ambiguous genitalia), and skeletal system (syndactyly, preaxial polydactyly, absent thumbs). Other common features included anemia/pancytopenia, premature graying of hair, and sensorineural hearing impairment. All patients presented severe NDD.

Upper panel: photographs showing phenotypic features of two patients (patients 27 and 35, Table 1). Patient 27, male index has low anterior hairline, thick scalp hair, thick eyebrows, synophrys, long eyelashes, long palpebral fissures, proptosis, strabismus, bulbous nose, low hanging columella, smooth philtrum, wide mouth, micrognathia, short neck, brachydactyly, right preaxial polydactyly, and bilateral syndactyly of the second and third toes. Patient 35, male index with coarse face, broad eyebrows, long palpebral fissures, wide mouth, thin vermilion of the upper lip, and bilateral absent thumbs. He presented generalized hypotonia and was severely emaciated. The patient deceased at 9 months old. Lower panel: summarized family trees of patients with ZNF699 homozygous LoF variants and clinical features. Patients presented with a severe phenotype that included congenital heart defects, gastrointestinal (intestinal atresia, pyloric stenosis, GER, hepatosplenomegaly), genitourinary (renal hypoplasia, cryptorchidism, chordee, hypospadias, ambiguous genitalia), and skeletal abnormalities (preaxial polydactyly, absent thumbs, syndactyly). Other recurrent features were generalized hypotonia, sensorineural hearing impairment, and premature hair graying. All patients have severe neurodevelopmental delay (NDD).
The first patient identified was a 2-year-old female born preterm (32 weeks) to consanguineous parents (patient 26, Table 1, Fig. 3). She has a similarly affected sibling, who is also homozygote for the same ZNF699 variant. A clinical summary of the patients from three families is presented in the Supplementary Information (patients 26, 35, and 38, Table 1, Fig. 3). Despite the clear phenotypical similarity of the 13 patients identified with homozygous LoF variants in ZNF699, little is known about the function of this gene, which was initially described in Drosophila in a study of alcohol dependence.33 The gene encodes a large nuclear zinc-finger protein, suggesting a molecular role in nucleic acid binding.34
We also detected variants in known candidate genes that had insufficient published evidence supporting causality (and no OMIM associated phenotypes). Our current data provides further evidence supporting confirmation of 31 candidate genes in 56 patients with a wide range of clinical phenotypes. These include cases with syndromic and nonsyndromic forms of NDD/ID, ciliopathy, oral–facial–digital syndrome, cardiomyopathy, syndromic short stature, and skeletal dysplasia. The identified genes are APC2, CAP2, EIF3F, GYG2, IFT57, ITFG2, LGI3, NEK10, NRAP, PAPPA2, PPP1R13L, WIPI2, ZNF526 (autosomal recessive, X-linked inheritance, Supplementary table 1); AFF3, BCORL1, CHD6, CNOT1, CTR9, DMXL1, FRYL, KLF7, MYCBP2, NRXN2, PHF21a, RAB11a, RALA, SPEN, TAF4, TANC2, ZNF292, ZNF462 (autosomal dominant, de novo variants, Supplementary Table 2). Selected examples from this group are described in the following sections.
PAPPA2
Dauber et al. reported the finding of two homozygous variants (missense and frameshift) in two unrelated families, with several children having significant postnatal growth retardation, long thin bones, long fingers and toes, mild microcephaly, abnormal dentine and teeth enamel, and mild dysmorphisms. In vitro analyses demonstrated that both variants caused a complete absence of PAPPA2 proteolytic activity;35 however, no additional patients have been reported to date. We identified two novel homozygous nonsense variants in PAPPA2, in two patients with short stature and dysmorphic features with no evident NDD. The phenotype is highly similar to the previously reported cases supporting a causal role of PAPPA2 in a novel short stature syndrome.
TAF4
A heterozygous de novo variant (frameshift) was reported in TAF4 by Kosmicki et al., in a patient with autism.36 The gene has no phenotypic association in OMIM (accessed 12 October 2020). Within this study, we identified two additional de novo LoF variants (splicing and nonsense) in two unrelated patients with dysmorphic features and NDD. TAF4 is highly intolerant to LoF as documented in gnomAD (pLi = 1). Expression of TAF4 varies during development and in the processes of cell differentiation; TAF4 is detected in various regions of the human brain, and it is believed to control the differentiation of human neural progenitor cells having a role in the regulation of neural development and brain function.37 The current data suggests that TAF4 haploinsufficiency leads to NDD in humans.
RAB11a
Hamdan et al. described three patients with developmental and epileptic encephalopathy as well as de novo missense variants in the RAB11a gene.38 We identified two additional variants in the same GTPase region of RAB11a in patients with microcephaly, NDD, and specific brain abnormalities. Dendritic spines are postsynaptic protrusions at excitatory synapses that are critical for proper neuronal synaptic transmission. RAB11a is part of the cascade controlling spine formation and function.39 When combined, the genetic and functional data support a causative role of RAB11a for NDD with epileptic encephalopathy and microcephaly.
MYCBP2
This gene is not associated to any phenotype in OMIM (accessed 12 October 2020). Neale and Kosmicki et al.36,40 reported de novo missense and frameshift variants in patients with autism spectrum disorder after screening a large cohort of patients. Recently, Takahashi et al. identified two variants (one of them confirmed as arisen de novo) in two cases with uterovaginal aplasia with concomitant defects, such as renal, skeletal malformations, hearing defects, and rare cardiac and digital anomalies known as Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome.41 Within this study, we detected three additional de novo variants (one likely affecting splicing and two missense) in three patients with NDD, microcephaly, and seizures. One case presented bilateral bifid thumbs, talipes, and scoliosis, without vaginal or uterine anomalies (two female patients, both were prepubertal). Our results support a causal link of MYCBP2 de novo variants and ID/NDD.
DISCUSSION
The ACMG/AMP guidelines for the interpretation of genetic variants are restricted to genes with established causality in human diseases,12 while variants in genes for which this evidence is insufficient are considered genes of unknown significance (“research” or “candidate” genes).12 Therefore, in routine diagnostics, many genes are excluded during the filtering process of exome/genome data.
Clear guidelines should be established to identify, classify, and report variants located in candidate genes. Recently, Strande et al.42 proposed a comprehensive framework within the ClinGen initiative to evaluate relevant genetic and functional evidence supporting or contradicting gene–disease associations. The curation system covers gene variant evidence based on genetic data, and functional or experimental evidence. Gene-supporting evidence includes the identification of several unrelated patients and variants, and the absence of contradicting data (i.e., high variant frequency in controls).42 Experimental evidence comprises data on gene function, and cellular and animal models.42
As part of this study, we describe a patient-centered workflow implemented for cases with inconclusive or no genetic diagnosis after ES/GS. The process extends the search and evaluation to variants detected in genes of unknown significance. From these, we suggest six novel disease–gene associations. The findings are exclusively based on the analyses performed on our data repository, which enabled further identification of unrelated patients displaying similar phenotypes. As a follow-up, functional work is needed to confirm and to understand the disease mechanisms and related pathophysiology. This is particularly relevant for genes such as IPO8 and ZNF699, as little is known about their function. For both genes, the high number of affected individuals identified, the similarities of their phenotype, and the putative LoF nature of the homozygous variants detected are compelling evidence favoring a gene–disease association. Furthermore, our results support causality of 31 additional candidate genes. Following the ClinGen guidelines, these 31 gene–disease associations can be upgraded from having “limited” evidence to genes with “moderate” or “strong” evidence, based on 56 patients.
Traditionally, discovery of novel gene–disease associations has been done by research labs; however, with this work, we show the enormous potential of diagnostic labs to uncover and validate candidate genes. Multiple strategies can be implemented to help identify novel disease genes, which will ultimately benefit the patients and families with rare genetic diseases. Genomic data analysis beyond known disease genes can be implemented in a routine diagnostic approach, as shown within this study. Finally, for genetic labs, reporting of variants in diagnostic genes versus candidate genes should be clearly differentiated since clinical validity is restricted to the former. Communication with referring physicians are critical for follow-up and further validation of the gene–disease associations.
In conclusion, our work shows the benefits of performing extended ES/GS analyses in patients with no genetic diagnosis combined with further data repository mining. Dedicated analyses of such data repositories that combine clinical and genetic information can be routinely performed to identify and confirm candidate genes. Genetic laboratories should be encouraged to pursue such analyses for the benefit of undiagnosed patients and their families.
Data availability
The data set that was generated and/or analyzed as part of this study is available from the corresponding author.
References
Clark, M. M. et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom. Med. 3, 16 (2018).
Trujillano, D. et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur. J. Hum. Genet. 25, 176–182 (2017).
Boycott, K. M. et al. International cooperation to enable the diagnosis of all rare genetic diseases. Am. J. Hum. Genet. 100, 695–705 (2017).
Ott, J., Wang, J. & Leal, S. M. Genetic linkage analysis in the age of whole-genome sequencing. Nat. Rev. Genet. 16, 275–284 (2015).
Alkuraya, F. S. Discovery of mutations for Mendelian disorders. Hum. Genet. 135, 615–623 (2016).
Hansen, A. W. et al. A genocentric approach to discovery of Mendelian disorders. Am. J. Hum. Genet. 105, 974–986 (2019).
Kaplanis, J. et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 586, 757–762 (2020).
Bertoli-Avella, A. M. et al. Successful application of genome sequencing in a diagnostic setting: 1007 index cases from a clinically heterogeneous cohort. Eur. J. Hum. Genet. 29, 141–153 (2021).
Bauer, P. et al. Development of an evidence-based algorithm that optimizes sensitivity and specificity in ES-based diagnostics of a clinically heterogeneous patient population. Genet. Med. 21, 53–61 (2019).
Bertoli-Avella, A. M. et al. Successful application of genome sequencing in a diagnostic setting: 1007 index cases from a clinically heterogeneous cohort. Eur. J. Hum. Genet. 29, 141–153 (2020).
den Dunnen, J. T. et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum. Mutat. 37, 564–569 (2016).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Trujillano, D. et al. A comprehensive global genotype-phenotype database for rare diseases. Mol. Genet. Genomic Med. 5, 66–75 (2017).
Massadeh, S. et al. ADAMTS19-associated heart valve defects: novel genetic variants consolidating a recognizable cardiac phenotype. Clin. Genet. 98, 56–63 (2020).
Shao, D. D. et al. A recurrent, homozygous EMC10 frameshift variant is associated with a syndrome of developmental delay with variable seizures and dysmorphic features. Genet. Med. https://doi.org/10.1038/s41436-021-01097-x (2021).
Holt, R. J. et al. De novo missense variants in FBXW11 cause diverse developmental phenotypes including brain, eye, and digit anomalies. Am. J. Hum. Genet. 105, 640–657 (2019).
Salpietro, V. et al. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat. Commun. 10, 3094 (2019).
Rehman, A. U. et al. Biallelic loss of function variants in PPP1R21 cause a neurodevelopmental syndrome with impaired endocytic function. Hum. Mutat. 40, 267–280 (2019).
Dulovic-Mahlow, M. et al. De novo variants in TAOK1 cause neurodevelopmental disorders. Am. J. Hum. Genet. 105, 213–220 (2019).
Guo, H. et al. Disruptive mutations in TANC2 define a neurodevelopmental syndrome associated with psychiatric disorders. Nat. Commun. 10, 4679 (2019).
Chivukula, R. R. et al. A human ciliopathy reveals essential functions for NEK10 in airway mucociliary clearance. Nat. Med. 26, 244–251 (2020).
Setty, S. R. et al. BLOC-1 is required for cargo-specific sorting from vacuolar early endosomes toward lysosome-related organelles. Mol. Biol. Cell. 18, 768–780 (2007).
Scott, I., Wang, L., Wu, K., Thapa, D. & Sack, M. N. GCN5L1/BLOS1 links acetylation, organelle remodeling, and metabolism. Trends Cell. Biol. 28, 346–355 (2018).
Zhang, A. et al. Biogenesis of lysosome-related organelles complex-1 subunit 1 (BLOS1) interacts with sorting nexin 2 and the endosomal sorting complex required for transport-I (ESCRT-I) component TSG101 to mediate the sorting of epidermal growth factor receptor into endosomal compartments. J. Biol. Chem. 289, 29180–29194 (2014).
Cheli, V. T. et al. Genetic modifiers of abnormal organelle biogenesis in a Drosophila model of BLOC-1 deficiency. Hum. Mol. Genet. 19, 861–878 (2010).
Loeys, B. L. et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 355, 788–798 (2006).
Lindsay, M. E. et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 44, 922–927 (2012).
Bertoli-Avella, A. M. et al. Mutations in a TGF-beta ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 65, 1324–1336 (2015).
van de Laar, I. M. et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 43, 121–126 (2011).
Micha, D. et al. SMAD2 mutations are associated with arterial aneurysms and dissections. Hum. Mutat. 36, 1145–1149 (2015).
Tao, G., Levay, A. K., Gridley, T. & Lincoln, J. Mmp15 is a direct target of Snai1 during endothelial to mesenchymal transformation and endocardial cushion development. Dev. Biol. 359, 209–221 (2011).
Wu, Z. Q. et al. A Snail1/Notch1 signalling axis controls embryonic vascular development. Nat. Commun. 5, 3998 (2014).
Scholz, H., Franz, M. & Heberlein, U. The hangover gene defines a stress pathway required for ethanol tolerance development. Nature. 436, 845–847 (2005).
Fagerberg, L. et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13, 397–406 (2014).
Dauber, A. et al. Mutations in pregnancy-associated plasma protein A2 cause short stature due to low IGF-I availability. EMBO Mol. Med. 8, 363–374 (2016).
Kosmicki, J. A. et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat. Genet. 49, 504–510 (2017).
Kazantseva, J., Tints, K., Neuman, T. & Palm, K. TAF4 controls differentiation of human neural progenitor cells through hTAF4-TAFH activity. J. Mol. Neurosci. 55, 160–166 (2015).
Hamdan, F. F. et al. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am. J. Hum. Genet. 101, 664–685 (2017).
Nishino, H. et al. The LMTK1-TBC1D9B-Rab11a cascade regulates dendritic spine formation via endosome trafficking. J. Neurosci. 39, 9491–9502 (2019).
Neale, B. M. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 485, 242–245 (2012).
Takahashi, K. et al. Exome and copy number variation analyses of Mayer-Rokitansky-Kuster- Hauser syndrome. Hum. Genome Var. 5, 27 (2018).
Strande, N. T. et al. Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the Clinical Genome Resource. Am. J. Hum. Genet. 100, 895–906 (2017).
Author information
Authors and Affiliations
Contributions
A.M.B.-A.: conceptualization, data curation and formal analysis, as well as writing of the original draft. K.K.K. and S.K.: conceptualization, data curation, analysis of NGS data and formal analysis. N.O.-H., K.T., C.B., M.E.R., A.U., R.H., A.L., R.A.-A., N.A.: data curation and analysis of NGS data. N.A., A.R., P.B.: review and editing of the manuscript. O.M., A.A., M.A., A.F.A., Z.A., M.A.A., F.H., Z.H., N.M.G.A., L.A., H.C., M.S.Z., I.H., A.A., W.E., F.M., M.A., F.S.A., N.A.A., A.M.A.: consulted and referred the patients and provided clinical data. All authors approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
A.M.B.-A., K.K.K., S.K., N.O.-H., N.O.-H., K.T., C.B., M.E.R., A.U., R.H., A.L., R.A.-A., V.K., N.A., and P.B. are employees of CENTOGENE GmbH. A.R. is the former CEO of CENTOGENE GmbH. The other authors declare no competing interests.
Ethics declaration
The current project has been conducted within a diagnostic setting and in a second step, utilized de-identified data and samples, and thus did not require institutional review board (IRB) approval in our jurisdiction. Informed consents were obtained, including specific consents for publication of patient images. The form contains a section for consent for genetic testing related to the disease(s) of the patient and consent for research (related to the main concern, but implicating genes not yet associated to human diseases). Additionally, the consent declaration included information regarding storage of the data and further processing for research purposes. The informed consent form is available in English and several other languages at https://www.centogene.com/downloads.html.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bertoli-Avella, A.M., Kandaswamy, K.K., Khan, S. et al. Combining exome/genome sequencing with data repository analysis reveals novel gene–disease associations for a wide range of genetic disorders. Genet Med 23, 1551–1568 (2021). https://doi.org/10.1038/s41436-021-01159-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-021-01159-0
This article is cited by
-
Facing the challenges to shorten the diagnostic odyssey: first Whole Genome Sequencing experience of a Colombian cohort with suspected rare diseases
European Journal of Human Genetics (2024)
-
Messenger RNA transport on lysosomal vesicles maintains axonal mitochondrial homeostasis and prevents axonal degeneration
Nature Neuroscience (2024)
-
KIF26A is mutated in the syndrome of congenital hydrocephalus with megacolon
Human Genetics (2023)
-
Reanalysis of exome sequencing data reveals a treatable neurometabolic origin in two previously undiagnosed siblings with neurodevelopmental disorder
Neurological Sciences (2023)
-
A founder DBR1 variant causes a lethal form of congenital ichthyosis
Human Genetics (2023)
