
Abstract
Purpose
We previously developed Haploseek, a method for comprehensive preimplantation genetic testing (PGT). However, some key features were missing, and the method has not yet been systematically validated.
Methods
We extended Haploseek to incorporate DNA from embryo grandparents and to allow testing of variants on chromosome X or in regions where parents share common haplotypes. We then validated Haploseek on 151 embryo biopsies from 27 clinical PGT cases. We sequenced all biopsies to low coverage (0.2×), and performed single-nucleotide polymorphism (SNP) microarray genotyping on the embryos’ parents and siblings/grandparents. We used the extended Haploseek to predict chromosome copy-number variants (CNVs) and relevant variant-flanking haplotypes in each embryo. We validated haplotype predictions for each clinical sample against polymerase chain reaction (PCR)-based PGT case results, and CNV predictions against established commercial kits.
Results
For each of the 151 embryo biopsies, all Haploseek-derived haplotypes and CNVs were concordant with clinical PGT results. The cases included 17 autosomal dominant, 5 autosomal recessive, and 3 X-linked monogenic disorders. In addition, we evaluated 1 Robertsonian and 2 reciprocal translocations, and 17 cases of chromosome copy-number counting were performed.
Conclusion
Our results demonstrate that Haploseek is clinically accurate and fit for all standard clinical PGT applications.
Similar content being viewed by others


INTRODUCTION
Preimplantation genetic testing (PGT) has been available for >20 years to help prevent the birth of children with Mendelian (monogenic) disorders and/or structural pathogenic variations.1 In PGT, embryos are generated by in vitro fertilization (IVF), and a biopsy from each embryo is genetically tested for monogenic disease (PGT-M), translocation or subchromosome copy-number imbalance (PGT-SR), and/or genome-wide aneuploidy (PGT-A).2 In Israel, PGT-M/SR has had a significant impact on families carrying pathogenic variants, due to relatively high uptake of genetic screening.3 Furthermore, part of the population will not interrupt a pregnancy for religious reasons.4,5 However, the current implementation of the method, based on amplifying and genotyping polymorphic markers flanking the pathogenic variant, is laborious, expensive, and requires long family-specific preparation.6 In addition, this technique limits the number of markers to be tested and does not easily facilitate embryo fingerprinting. Another limitation is that informative markers surrounding the relevant variant are not always available for consanguineous couples, as markers are often identical in both partners. In Israel and generally in the Middle East, there is a high number of consanguineous unions in certain populations.7
The fast developments in whole-genome amplification (WGA) and genotyping/sequencing technologies, together with innovative statistical modeling of single-cell DNA sequencing, have led to a wave of recent publications offering fast and accurate technologies that can simultaneously perform PGT-A, PGT-SR, and PGT-M.6,8,9,10,11,12,13,14,15,16,17 In most implementations, it is assumed that the genome of an affected family member is available, in order to tease apart, or “phase,” the two haplotypes of each parent, and thereby identify which haplotype (the one carrying the disease variant vs. the wild-type haplotype) was transmitted to each embryo.
We recently developed a PGT method termed Haploseek,18 which relies on microarray genotyping of the parents and an affected child, along with very low–coverage whole-genome sequencing of the embryos. Haploseek is an accurate clinical grade method that can reach a turnaround time of 24 hours at an affordable cost.18 To overcome missing genotypes due to the shallow sequencing coverage, we used statistical modeling via hidden Markov models (HMMs) for inferring the chromosome transmitted from each parent and the copy number at each autosomal region in each embryo. However, the original implementation of this method did not account for PGT cases where a disease-affected child was unavailable for haplotype phasing reference. Moreover, X-linked disease diagnosis required manual entry of X chromosome ploidy for each embryo under consideration. In addition, for consanguineous couples, haplotype prediction accuracy was somewhat compromised within genomic regions with extensive haplotype sharing.
Here, we report a substantial expansion and validation of the capabilities of Haploseek, by clinical testing in a cohort of 151 embryos from 27 couples. To improve the applicability of Haploseek, we extended it to include three new functionalities. First, we demonstrate practical diagnosis of embryos from consanguineous couples using tailored statistical modeling. Second, our method can now handle families where one or more embryo grandparents (parents of the couple performing PGT) are available instead of an affected child. We used the grandparents for phasing the haplotypes of the parents, and then used that phasing information to accurately diagnose the embryo. Third, we added functionality for X-linked disorder testing, automatically accounting for sex-specific X chromosome ploidy.
We demonstrate successful clinical validation with 100% accuracy of Haploseek across a broad selection of single gene and copy-number variant disorders.
MATERIALS AND METHODS
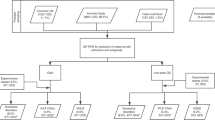
An overview of the evaluation strategies
We used two approaches to evaluate the results of our methods.
To evaluate the performance of Haploseek in consanguineous families and for the initial evaluation of the X chromosome method, we used low-depth sequencing of single-cell DNA from tissue cultures as described in our previous paper,18 with key points also mentioned in various subsections below.
The remaining evaluations were performed on residual genetic material donated for research from embryos that underwent PGT due to various conditions. In this setting, Haploseek‘s results for the predicted genotype of each embryo were compared against clinical PGT-M results based on polymerase chain reaction (PCR) amplification of flanking markers. Haploseek’s predicted copy-number status was validated by testing the same WGA material with an established commercial kit for single-cell copy-number variant (CNV) detection (VeriSeq-PGS; Illumina).
Sample processing pipeline for grown children
Genomic DNA of parents and children from four families was collected as described in our previous paper.18 To emulate a PGT pipeline, we prepared lymphoblast single cells for the children, also as previously described.18
Sample processing pipeline for PGT embryos
DNA samples were collected from PGT couples and their family members during routine pre-PGT workup. Embryo biopsies were all processed for WGA during the course of clinical PGT cycles. Residual WGA material was utilized in this study.
Single-nucleotide polymorphism (SNP) microarray analysis
Genomic DNA was extracted from peripheral blood using the FlexiGene DNA kit (Qiagen). Subsequently, DNA samples were subject to CytoScan® 750K or CytoScan® HD SNP array (Affymetrix) and then scanned on an Affymetrix GeneChip (GCS3000) Scanner. Chromosome Analysis Suite (Chas; Affymetrix) was used to extract genotype calls and copy-number predictions (where relevant) from each array.
Whole-genome amplification and low-coverage genome sequencing
Single culture cell/blastomere/blastocyst DNA underwent WGA using the PicoPLEX WGA kit (Rubicon Genomics) according to the manufacturer’s protocol. Amplified DNA from each cell was then converted into genome sequencing libraries using the Nextera XT library prep kit (Illumina). Resultant libraries were normalized and pooled for 1×75 single read or 2×75 paired end sequencing on a NextSeq 500 (Illumina) instrument to generate 0.2–0.4× genomic sequence coverage per sample.
NGS variant calling
Low-coverage high-throughput sequencing reads were aligned to the reference human genome (hg19) using Burrows–Wheeler alignment (BWA),19 and the reads mapping to reference and alternate alleles were counted using samtools mpileup at SNP positions matching those on the Affymetrix arrays.
Haplotype prediction using low-coverage sequencing data
Our data include low-coverage sequencing reads for each child or embryo, and microarray genotypes for the two parents and a child reference or one or more embryo grandparents. The computational pipeline for haplotype prediction involving a child reference was described in our previous paper.18 For haplotype predictions involving a grandparent(s) reference and X chromosome phasing, see Supplementary Materials and Methods.
Copy-number variant prediction
CNV prediction was performed as we previously described.18
Output visualization
The user-friendly interface in18 was used to visualize data output on a web browser in two plots depicting the haplotype prediction (Viterbi path) and the “marginal” (posterior) probabilities, respectively.
Regions of consanguinity
Here, we consider the setting when the diagnosis of embryos is based on the genomes of parents and an affected child, and the goal is to determine, for each embryo, whether its maternal/paternal haplotypes are the same as in the affected child or not. In the case of consanguineous families (i.e., the parents are related), it can be difficult to infer the origin of some embryo haplotypes. For prioritization of embryos from consanguineous families carrying autosomal recessive pathogenic variants, we provide additional information. We defined regions of consanguinity (ROCs) as genomic regions where the parents had identical heterozygous genotypes. Specifically, we designated any genomic region as a ROC if, for every set of 200 consecutive array SNPs in the region, no more than 95% of the paternal genotypes were homozygous and at least 95% of the father’s and mother’s genotypes were identical. In these regions, in addition to plotting the posterior probability of inheritance of each haplotype from each parent, we also computed a “diff” score, indicating the probability that the embryo has a genotype different by at least one haplotype from the affected child (i.e., the embryo is either heterozygous or homozygous to the wild-type haplotype). To compute the score, we simply summed over all hidden states in our Markov chain model that are consistent with the embryo having a different genotype compared to the affected child. (For a description of our HMM, see our previous paper.18) Visually, in these regions we provide a third plot, which we term “diff plot,” illustrating the probability that the examined embryo is not homozygous to the disease haplotype.
RESULTS
An overview of our results
We performed clinical validation of Haploseek on a cohort of 151 embryo biopsies from 27 different families altogether. Applications included PGT-M, PGT-A, and/or PGT-SR, and Haploseek test results for each biopsy were 100% concordant with clinical PCR-based PGT-M and with VeriSeq-PGS-based PGT-A/PGT-SR test results (see below).
For expanded clinical application of Haploseek, the following key features were added: (1) enhanced accuracy for recessive disease diagnosis in embryos from consanguineous couples, (2) support for parental haplotype phasing using microarrays from embryo grandparents instead of disease-affected siblings of the embryos, and (3) X-linked disease diagnosis. In the following, we describe the results of our experiments for validation of our approach.
Addressing autosomal recessive disease diagnosis in consanguineous families
Our clinical laboratory is located in a geographic region in which consanguineous unions occur with high incidence.7 Consequently, consanguineous couples often seek molecular PGT-M services in our lab. Nonetheless, PGT-M for these couples is not trivial because of extensive haplotype sharing among the partners, which creates a challenge not only for Haploseek but also for other comprehensive genome-wide PGT strategies13,20 and gold standard PCR-based molecular testing methods. Previously, we reported compromised haplotype prediction accuracy when applying Haploseek to embryos from consanguineous families.18 Most errors occurred in long regions of the genome where the two parents shared both of their haplotypes, i.e., both parents had haplotype pairs H1H2, where H1 denotes the haplotype that carries a recessive disease variant and H2 is a wild-type haplotype. In these cases, the parents are said to be “IBD2,” where IBD stands for “identical-by-descent,” and 2 means that both haplotypes are shared. Herein,18 we call these regions ROCs. Within ROCs, it is difficult to determine which haplotype was transmitted from each parent, in particular when the embryo is heterozygous (H1H2). However, the question of exactly which parent has transmitted the mutant haplotype is inconsequential when performing PGT-M for an autosomal recessive disease. As long as the embryo is not a homozygous mutant (H1H1), the embryo can be implanted. In other words, conclusive detection of at least one parental wild-type allele in the embryo (even if it is not clear which parent/s has transmitted the allele/s) is sufficient grounds to prioritize the embryo for transfer.
Using the aforementioned guiding principles, we added to Haploseek the ability to diagnose the presence or absence of homozygous mutant alleles in consanguineous families. In this setting, the DNA of the parents and an affected child (“child 1”) were genotyped on a microarray prior to sequencing the embryos to low depth. Then, we added a ROC identification feature to the analysis module, whereby all parental arrays in each Haploseek cycle are scanned for ROCs and each ROC is flagged accordingly. The ROCs are highlighted on the marginal plots of the user interface (Fig. 1; green dots) so as to draw the attention of the medical practitioner during PGT-M result reporting. Then, to reach clinical diagnosis of the embryo in each ROC, we show a diff plot, which evaluates the probability that the embryo is genetically identical to the homozygous affected child 1 sibling or “different” (meaning either heterozygous or homozygous wild type; Fig. 2). On the user interface, the diff plot is easily accessed by clicking a checkbox at the top the screen.

Pictured is a screen capture of chr9 haplotype data from children 2 and 3 in consanguineous family 2. The samples were derived from single cells sequenced to depth 0.2×. Maternal and paternal haplotypes are each depicted in prediction and marginal plots for each child. The prediction plot indicates the hidden Markov model (HMM) haplotype prediction (the Viterbi path) in child 2 or child 3, as indicated, relative to the affected child 1 reference (not depicted). Dark red and dark blue shaded segments indicate that child 2/3 matches child 1’s maternal and paternal haplotypes, respectively. Lighter shading indicates that child 2/3 matches the wild-type haplotypes that are not present in child 1. The marginal (posterior probability) plot indicates the degree of confidence with which the HMM is reporting a certain haplotype prediction. A marginal value of 1 means a high confidence prediction of a child 1 matching haplotype. A marginal value of 0 means high confidence prediction of a child 1 mismatching haplotype. We call sites with marginal values between 0.01 and 0.99 low confidence. These sites mostly appear near meiotic recombination sites. The marginal probabilities are plotted as dots at the single-nucleotide polymorphism (SNP) sites on the arrays that were also successfully sequenced. Green dots flag a region of consanguinity (ROC), in which the parents share both of their haplotypes (and those haplotypes are not identical to one another). Note that there is a single ROC that extends over the centromere (the centromere appears as a thin black line with no sequenced array SNPs toward the middle of the chromosome). The “show diff” checkbox on the top left is unchecked, thereby indicating that conventional Haploseek haplotype predictions are being viewed. The plot suggests two crossovers in the mother, within the ROC, upstream and downstream of the centromere in both child 2 and child 3, with low-confidence marginal scores. Thus, from this plot alone it is impossible to determine whether the mother’s paternal or maternal haplotype was transmitted to the embryo in close proximity of the maternal crossover sites. However, as long as the paternal wild-type haplotype was transmitted, the embryo could be transferred. The purple rectangle indicates an evaluation region where child 1 is H1H1 and both parents are H1H2. Regions such as these are best interpreted in the diff plot (see Fig. 2).

The diff plot depiction of chr9 from the same children as in Fig. 1. This plot simplifies the diagnosis of autosomal recessive disease in consanguineous families. Results are summarized as “homozygous for pathogenic haplotype,” meaning that child 2/3 is identical in both haplotypes to child 1, or “wild type or heterozygous” meaning that child 2/3 is unlike child 1 in at least one haplotype, and thus should be prioritized for transfer in preimplantation genetic testing for monogenic disease (PGT-M) cases. “Diff plot” indicates the degree of confidence with which the hidden Markov model is reporting a certain embryo classification. A diff plot value of 1 means a high-confidence prediction of a nondiseased embryo (i.e., different in at least one haplotype from child 1). A diff plot value of 0 means high-confidence prediction of a diseased embryo, fully matching child 1. We call sites with diff plot values between 0.01 and 0.99 low confidence. Green dots mark regions of consanguinity (ROCs) in which the parents share both of their haplotypes. The purple rectangle indicates the same evaluation region as in Fig. 1. Note that the “show diff” checkbox on the top left is checked, thereby indicating that diff plot embryo classification is being viewed.
We evaluated the performance of the diff plot diagnosis on two consanguineous families from our previous report.18 In these families, parents and grown children were available. Single cells were collected from a lymphoblast culture of each child and their DNA was amplified and sequenced, and genomic DNA from bulk blood cells was genotyped on microarrays. In clinically relevant ROCs (which we denote as “evaluation regions” in Table S1) where child 1 is H1H1 (i.e., more than 95% of the child 1 genotypes are homozygous) and both parents are H1H2, diff plot predictions were perfectly accurate on ROC SNPs that passed the confidence filter (compared with results from bulk DNA genotyping). For one child in family 2, the haplotype model had lower diagnostic yield than the diff plot model. In this and similar cases, as long as the diff plot confidence is high, a test result may be reported for the embryo if there is sufficient evidence to indicate that the embryo is or is not homozygous mutant like child 1.
In Figs. 1 and 2, we present a scenario in family 2 where the diff plot yields a clinically actionable result even in a case where both parental haplotypes cannot be resolved. In Fig. 1, we see a maternal recombination event inside an “evaluation region” in both child 2 and child 3, indicating that the child 1 reference was recombinant in this region. Hence the maternal marginal score is low confidence and her haplotype cannot be determined in both children. However, the paternal haplotypes in the same genomic region are clearly defined as "wild type" (child 1 mismatching) in child 2 and "mutant" (child 1 matching) in child 3. Looking at the diff plot in Fig. 2, we see that the marginal score for child 3 is poor, yet very high in child 2. This is because to child 3, the father had clearly transmitted his "mutant" haplotype, while the mother’s haplotype cannot be determined due to a crossover event. Homozygous "mutant" status cannot be differentiated from heterozygous state in this case. In contrast, child 2 had clearly inherited the father’s wild-type haplotype, thus even without discerning the maternal haplotype, we have sufficient evidence to “diagnose” child 2 because he/she definitely is not homozygous mutant like child 1 but rather homozygous wild type or heterozygous (which are both genetic states that are fit for embryo transfer). This delineates a rather simple case of how the diff plot can yield actionable results in samples such as child 2 even when complete haplotype predictions are not possible.
Clinical validation of Haploseek and utilization of embryo grandparent references for haplotype phasing
Haploseek was originally established on the assumption that SNP microarray genotypes are available for a living child, which could be used to phase the parental array genotypes. However, in the precase clinical setting, DNA from a living child of the couple is not always available, e.g., if the couple is childless. In fact, in our clinic, at least half of precase haplotype workup is performed with DNA from one or more parents of the parents (grandparents of the embryos) because child DNA is unavailable. Thus, given this limitation of Haploseek, we devised a new grandparent module to enable utilization of grandparent DNA in lieu of child DNA for phasing the parental genotypes.
Our new module allows the user to input 1 to 4 grandparent array genotypes as reference genomes for phasing. We redesigned our HMM to accommodate this change (Supplementary Materials and Methods). Briefly, our HMM has read counts as observations (with probabilities based on a beta-binomial model), and the hidden states represent which haplotype was transmitted from each parent (with transitions reflecting crossovers). The parental haplotypes are determined based on grandparental genotypes, taking into account possible genotyping and phasing errors. For heterozygous parental SNPs in which grandparents are uninformative or missing, we assign equal probability to each haplotype assignment. The prediction of which haplotype was transmitted to the embryo is then output in the user interface, labeled according to the source of each haplotype (maternal grandmother or grandfather and the paternal grandmother or grandfather; Fig. 3).

(a) A pedigree of family 24, in which the mother and maternal grandmother (of embryo 68e-1) are both heterozygous for a germline 0.5-Mb duplication on chr16. For haplotype phasing of the mother, the maternal grandmother’s DNA was genotyped. (b) DNA microarray analysis demarcates the location of the duplication on chr16 in both the mother and the maternal grandmother (genomic coordinates indicated on top). The panel shows the microarray analysis suite (Chas) screen capture of the chr16p duplication in both individuals. (c) Screen captures of Haploseek haplotype prediction plots for the familial chr16 duplication in embryo biopsy 68e-1 of family 24. The approximate location of the duplication is marked by a dashed vertical line. The plot shows haplotype predictions in the +/−2 Mb genomic region flanking the chr16 duplication. Overall, the results are depicted in similar fashion to that in Fig. 1, except that the haplotype predictions are defined with respect to grandparental reference (see legend on the top right), as opposed to the child 1 reference. The pictured sample exhibits a high-confidence maternal grandfather haplotype at the familial pathogenic variant site. Since the maternal grandfather does not carry the chr16 duplication, the embryo is wild type. This result and all other clinical applications of Haploseek using grandparent references (summarized in Table S2) were validated by polymerase chain reaction (PCR)-based PGT-M. Note that paternal grandparents were not genotyped, and hence the paternal haplotype of the embryo is equally likely to be grandpaternal or grandmaternal (marginal probability equals 0.5 for the entire paternal haplotype and therefore the paternal haplotype prediction should be ignored).
To evaluate the accuracy of our grandparents module, we ran Haploseek on 16 different clinical PGT-M cases in which one or more grandparents were used as the phasing reference. The results are presented in Table S2, covering various autosomal recessive and dominant diseases, and 73 embryos overall. The results for all embryos were completely concordant with standard PCR-based PGT-M test on the same embryo biopsies.
X-linked disease testing
Haploseek provides accurate whole-autosome haplotype predictions.18 However, the underlying statistical model did not take the X chromosome ploidy into account, and the accuracy for X-linked diseases was not established. We added a new X chromosome module to Haploseek, which evaluates which maternal X chromosome was transmitted to the embryo. We implemented this feature for both child-based and grandparent-based phasing pipelines (Fig. 4).

(a) A pedigree of family 15, in which the mother and maternal grandmother (of embryo 101Q-2) are both heterozygous for a premutation allele at the FMR1 gene locus on chrX. For haplotype phasing of the mother, the maternal grandmother’s DNA was genotyped. (b) Screen captures of Haploseek haplotype prediction plots for the familial FMR1 premutation allele in embryo biopsy 101Q-2 of family 15. The approximate location of the FMR1 gene is marked by a dashed vertical line. Results are depicted as in Fig. 3c, except that the haplotype predictions are for the +/−2 Mb flanking region of the FMR1 gene on chrX. The pictured sample exhibits a high-confidence maternal grandfather haplotype at the family premutation site. Since the maternal grandfather does not carry the FMR1 premutation, the embryo is wild type. This result and all other clinical applications of Haploseek for X-linked diseases (summarized in Table S4) were validated by polymerase chain reaction (PCR)-based PGT-M. Note that Haploseek does not output paternal haplotype predictions on chrX because male embryos do not have a paternal chrX and female embryos have a known non-PAR paternal chrX haplotype. Hence, the marginal probability is marked on the plot as 0.5 for the entire paternal haplotype and the paternal haplotype prediction should be ignored.
In initial testing, we determined genome-wide accuracy of X chromosome haplotypes in three families of parents and grown children, as we used for the evaluation of the consanguinity module. Table S3 shows very high per-SNP accuracy of this module, provided that haplotype predictions are determined in nonpseudoautosomal regions of chrX. To further evaluate the clinical utility of the X chromosome module, we performed X-linked Haploseek on four different clinical PGT-M cases (three cases with a child phasing reference and one with grandparent phasing references). Here too, the Haploseek X chromosome haplotype predictions on 11 embryos overall were completely concordant with standard PCR-based PGT-M test results on the same embryo biopsies (Table S4). Hence, we conclude that Haploseek is well suited for clinical X-linked disease PGT-M.
Evaluation of Haploseek on additional families
To further evaluate the clinical utility of Haploseek, we tested another seven PGT-M cases using child DNA as the phasing reference. Like the grandparent families above, Haploseek showed complete concordance with PCR-based PGT-M in all 67 embryos for various autosomal recessive or dominant disorders (Table S5). Thus, we demonstrate that Haploseek performs well in clinical case testing regardless of whether child or grandparent DNA is used as the haplotype phasing reference.
Expanded clinical validation of Haploseek for PGT-A or PGT-SR applications
One of the primary benefits to Haploseek is the ability to diagnose PGT-A and PGT-SR targets in the same diagnostic test and same embryo biopsy as that of PGT-M. However, extensive clinical validation data for PGT-A and PGT-SR applications of Haploseek was lacking. Here, we validated clinical PGT-A/SR applications for all aforementioned cases where clinical PGT-A/SR results were available (Tables S2, S4, and S5). Our results show that Haploseek PGT-A/SR results were completely concordant with that of VeriSeq-PGS on 72/72 embryos where a result was obtained from both methods.
DISCUSSION
We report here on the implementation and validation of extensive improvements to the Haploseek analytical pipeline on a large cohort of 151 embryos. Our results demonstrate that Haploseek is highly accurate, and is thus fit for all clinical PGT applications, whether it be PGT-A, PGT-SR, or PGT-M.
For PGT-M, we have now addressed prior deficiencies in the prototypical version of Haploseek. Adding the option to use the embryo’s grandparents as phasing references means that the implementation of Haploseek now covers most PGT-M cases. In 2019, 203 of 208 families (97.6%) who performed PGT-M in our clinic also provided DNA from at least one affected child or grandparent. The remaining families presented with either de novo pathogenic variants in one of the parents, or children/parents of the couple were not available. Hence, Haploseek is on par with other genome-wide comprehensive PGT methods such as karyomapping and OnePGT.8,21 However, it would still be important to adapt Haploseek and similar comprehensive PGT methods for parents-only embryo haplotype phasing, especially in the context of de novo pathogenic variant PGT. Indeed, various studies have already put forward some suggestions to address the limitations of relying on family member reference DNA for PGT.16,17,22,23
Another matter that has not been adequately addressed by existing comprehensive PGT methods is consanguinity. In previous reports involving karyomapping as the PGT method, consanguinity accounted for all misdiagnosed embryos.13,24 Moreover, OnePGT explicitly disclaims any use of their platform for consanguineous unions.20 Hence, the establishment of a comprehensive PGT method with a clinically valid solution for consanguineous families is necessary. In this report, we describe a new feature of the Haploseek platform that accurately discriminates homozygous recessive disease-affected embryos from healthy ones in regions where parents have identical pairs of haplotypes. We show here that embryo prioritization decisions in such cases are highly reliable. Thus, Haploseek should be considered as a viable comprehensive method for consanguineous couples seeking PGT-M for autosomal recessive disorders. Certainly, in regions such as the Middle East, where consanguineous unions are relatively common, the prospect of a universal assay for all recessive disorders is an attractive alternative to existing PGT solutions.
The following are limitations of this study. Although molecular test results from Haploseek were cross-checked by orthogonal methods on the same embryo biopsies, clinical outcome data were not available for each embryo at the time of manuscript preparation. While follow up for pathogenic variant-bearing biopsies is not possible given that such embryos are not transferred in clinical PGT cases, pregnancy outcome from embryos that otherwise would be prioritized for transfer (based on genetic testing) was not evaluated in this report. Likewise, live birth rate for embryo transfers leading to pregnancy was not calculated because this study was prepared before all pregnancies reached full term. Thus, our findings are limited to molecular test accuracy, and future studies will be necessary to assess the effect of the method on pregnancy and “take-home baby” outcomes.
Thus, to summarize, we have demonstrated that Haploseek is universally applicable for molecular testing of all types of inherited diseases, is highly accurate, and can also be successfully applied for consanguineous couples. The mission of our Haploseek platform is to offer affordable comprehensive and universal PGT. Haploseek accomplishes this mission by enhancing PGT throughput for midsized laboratories with access to microarray and high-throughput sequencing platforms. Haploseek does not require more than 0.2× mean genome coverage per sequenced embryo, making it one of the most affordable PGT-M sequencing-based solutions currently available. Nonetheless, we acknowledge that dependence on array genotyping of the parents and a family member reduces accessibility in some laboratories. Ideally, we would thus replace array genotyping of parents and family members with low-pass genome sequencing. This could save costs and simplify the analytical pipeline, as parents, relatives, and embryos would be jointly sequenced. However, this will require further research and development, and the anticipated cost saving may become a reality only upon further reduction of sequencing costs.
Data availability
De-identified source data for this study are available upon request.
References
Handyside, A. H., Kontogianni, E. H., Hardy, K. & Winston, R. M. Pregnancies from biopsied human preimplantation embryos sexed by Y-specific DNA amplification. Nature. 344, 768–770 (1990).
Simpson, J. L., Kuliev, A. & Rechitsky, S. Overview of preimplantation genetic diagnosis (PGD): historical perspective and future direction. Methods Mol. Biol. 1885, 23–43 (2019).
Zlotogora, J. The Israeli national population program of genetic carrier screening for reproductive purposes How should it be continued?. Israel J. Health Policy Res. 8, 73 (2019).
Zuckerman, S., Gooldin, S., Zeevi, D. A. & Altarescu, G. The decision-making process, experience, and perceptions of preimplantation genetic testing (PGT) users. J. Assist. Reprod. Genet. 37, 1903–1912 (2020).
Zuckerman, S., Zeevi, D. A., Gooldin, S. & Altarescu, G. Acceptable applications of preimplantation genetic diagnosis (PGD) among Israeli PGD users. Eur. J. Hum. Genet. 25, 1113–1117 (2017).
Vermeesch, J. R., Voet, T. & Devriendt, K. Prenatal and pre-implantation genetic diagnosis. Nat. Rev. Genetics 17, 643–656 (2016).
Bittles, A. H. & Black, M. L. Evolution in health and medicine Sackler colloquium: consanguinity, human evolution, and complex diseases. Proc. Natl. Acad. Sci. U. S. A. 107, 1779–1786 (2010).
Masset, H. et al. Multi-centre evaluation of a comprehensive preimplantation genetic test through haplotyping-by-sequencing. Hum. Reprod. 34, 1608–1619 (2019).
Zamani Esteki, M. et al. Concurrent whole-genome haplotyping and copy-number profiling of single cells. Am. J. Hum. Genet. 96, 894–912 (2015).
Yan, L. et al. Live births after simultaneous avoidance of monogenic diseases and chromosome abnormality by next-generation sequencing with linkage analyses. Proc. Natl. Acad. Sci. U. S. A. 112, 15964–15969 (2015).
Handyside, A. H. et al. Karyomapping: a universal method for genome wide analysis of genetic disease based on mapping crossovers between parental haplotypes. J. Med. Genet. 47, 651–658 (2010).
Xiong, L., Huang, L., Tian, F., Lu, S. & Xie, X. S. Bayesian model for accurate MARSALA (mutated allele revealed by sequencing with aneuploidy and linkage analyses). J. Assist. Reprod. Genet. 36, 1263–1271 (2019).
Natesan, S. A. et al. Genome-wide karyomapping accurately identifies the inheritance of single-gene defects in human preimplantation embryos in vitro. Genet. Med. 16, 838–845 (2014).
Murphy, N. M., Samarasekera, T. S., Macaskill, L., Mullen, J. & Rombauts, L. J. F. Genome sequencing of human in vitro fertilisation embryos for pathogenic variation screening. Sci. Rep. 10, 3795 (2020).
Treff, N. R. et al. Validation of concurrent preimplantation genetic testing for polygenic and monogenic disorders, structural rearrangements, and whole and segmental chromosome aneuploidy with a single universal platform. Eur. J. Med. Genet. 62, 103647 (2019).
Yuan, P. et al. A whole-genome sequencing-based novel preimplantation genetic testing method for de novo mutations combined with chromosomal balanced translocations. J. Assist. Reprod. Genet. 37, 2525–2533 (2020).
Chen, S., et al. Comprehensive preimplantation genetic testing by massively parallel sequencing. Hum. Reprod. 36, 236–247 (2021).
Backenroth, D. et al. Haploseek: a 24-hour all-in-one method for preimplantation genetic diagnosis (PGD) of monogenic disease and aneuploidy. Genet. Med. 21, 1390–1399 (2019).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 25, 1754–1760 (2009).
Agilent Technologies. Agilent OnePGT SolutionAn NGS-based, single workflow solution for PGT-M, PGT-SR and PGT-A. https://www.agilent.com/cs/library/usermanuals/public/G9425-90050.pdf (2020).
Gould, R. L. & Griffin, D. K. Karyomapping and how is it improving preimplantation genetics? Exp. Rev. Mol. Diagn. 17, 611–621 (2017).
Li, Q. et al. Haplotyping by linked-read sequencing (HLRS) of the genetic disease carriers for preimplantation genetic testing without a proband or relatives. BMC Med Genomics. 13, 117 (2020).
Zhang, S. et al. Long-read sequencing and haplotype linkage analysis enabled preimplantation genetic testing for patients carrying pathogenic inversions. J. Med. Genet. 56, 741–749 (2019).
Konstantinidis, M. et al. Live births following karyomapping of human blastocysts: experience from clinical application of the method. Reprod. Biomed. Online 31, 394–403 (2015).
Acknowledgements
The authors thank the Shaare Zedek Mirsky intramural grant for funding this research. S.C. and D.A.Z. thank the Hebrew University of Jerusalem Center for Interdisciplinary Data Science Research (CIDR; grant number 3035000322).
Author Information
Conceptualization: D.A.Z., D.B., S.C., G.A. Data curation: D.A.Z., F.Z. Formal analysis: D.A.Z., D.B., E.H.-S., S.Z., G.A. Funding acquisition: D.A.Z., P.R., S.C., G.A. Investigation: D.A.Z., D.B., E.H.-S., F.Z., R.S., S.Z., S.S., S.C., G.A. Methodology: D.A.Z., D.B, S.C., T.M., F.Z., S.Z. Resources: P.R., R.S., T.E.-G., I.B.-A., A.B.-Y. Software: D.B., F.Z. Supervision: D.A.Z., E.H.-S., P.R., R.S., I.B.-A., S.C., G.A. Validation: D.A.Z., E.H.-S., G.A. Visualization: F.Z.; Writing—original draft: D.A.Z.; Writing—review & editing: D.A.Z., D.B., S.C., G.A.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics Declaration
Ethical approval for this study was obtained from the Shaare Zedek Medical Center institutional review board. DNA, tissue culture samples, and human embryo biopsies in this study were donated to the Shaare Zedek Medical Genetics Institute for research with informed consent according to Shaare Zedek institutional review board guidelines and as set forth in the Declaration of Helsinki.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zeevi, D.A., Backenroth, D., Hakam-Spector, E. et al. Expanded clinical validation of Haploseek for comprehensive preimplantation genetic testing. Genet Med 23, 1334–1340 (2021). https://doi.org/10.1038/s41436-021-01145-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-021-01145-6
