
Abstract
Purpose
Azoospermia affects 1% of men and it can be the consequence of spermatogenic maturation arrest (MA). Although the etiology of MA is likely to be of genetic origin, only 13 genes have been reported as recurrent potential causes of MA.
Methods
Exome sequencing in 147 selected MA patients (discovery cohort and two validation cohorts).
Results
We found strong evidence for five novel genes likely responsible for MA (ADAD2, TERB1, SHOC1, MSH4, and RAD21L1), for which mouse knockout (KO) models are concordant with the human phenotype. Four of them were validated in the two independent MA cohorts. In addition, nine patients carried pathogenic variants in seven previously reported genes—TEX14, DMRT1, TEX11, SYCE1, MEIOB, MEI1, and STAG3—allowing to upgrade the clinical significance of these genes for diagnostic purposes. Our meiotic studies provide novel insight into the functional consequences of the variants, supporting their pathogenic role.
Conclusion
Our findings contribute substantially to the development of a pre–testicular sperm extraction (TESE) prognostic gene panel. If properly validated, the genetic diagnosis of complete MA prior to surgical interventions is clinically relevant. Wider implications include the understanding of potential genetic links between nonobstructive azoospermia (NOA) and cancer predisposition, and between NOA and premature ovarian failure.
Similar content being viewed by others



INTRODUCTION
Infertility is a multifactorial and heterogeneous condition affecting approximately 1 in 6 couples.1 The most severe form of male infertility is nonobstructive azoospermia (NOA) occurring in approximately 1% of all men.2 The etiology remains unknown in about 40% of NOA, and genetic factors are likely to play a major role. Although a growing number of fertility genes have been identified, the clinical impact of these findings is largely limited by the lack of validation in independent study populations.1,3 Hence, the identification of novel causes of NOA and the validation of previously reported genetic defects in independent cohorts are essential steps toward the development of diagnostic genetic tests.
Men affected by NOA might present with various testicular histologies, including Sertoli cell only (SCO), maturation arrest (MA) at different stages (such as spermatogonial and spermatocyte arrest [SGA, SCA]), and hypospermatogenesis. Complete MA is characterized by the absence of haploid germ cells and in this case even multiple and/or microsurgical biopsies for testicular sperm extraction ([m]TESE) to recover spermatozoa for subsequent in vitro fertilization will be unsuccessful. In contrast, in cases of incomplete MA, some tubules containing round or later stage spermatids may be found. Currently available clinical tools cannot distinguish between complete MA and other spermatogenic disturbances leading to azoospermia. Consequently, TESE is offered to all NOA patients as the only treatment option. The etiology of MA is poorly understood, and although genetic factors are predicted to play a relevant role, among the 13 MA genes reported in more than one patient3,4,5,6 only TEX11 variants7 and the complete AZFb deletion8 are currently considered clinically relevant. Hence, the development of a pre-TESE diagnostic NOA gene panel would be of high clinical benefit to prevent unnecessary surgery in patients with pure MA.
In this study, we successfully applied exome sequencing to uncover genetic causes in highly selected NOA men with MA, and we demonstrate a high clinical yield through the identification of novel MA genes as well as the validation of previously reported genes, all associated with TESE-negative NOA.
MATERIALS AND METHODS
Subjects
For the initial cohort, from a total of 1400 infertile men attending the Fundacio Puigvert, we selected 235 idiopathic NOA (iNOA) patients according to the following criteria: (1) absence of all known acquired (e.g., chemotherapy, bilateral cryptorchidism, testis torsion) and genetic (karyotype aberrations, Y-chromosomal AZF deletions) causes of azoospermia; (2) availability of DNA; and (3) stored testis biopsy. We further prioritized the cohort according to testis phenotype of SGA or SCA and family history (azoospermic brothers, infertile sisters, or consanguinity). The definition of the testis phenotype is based on multiple testis biopsies (one fragment is analyzed by the pathologist who described the histological picture, the remaining fragments by the embryologist who searched for spermatozoa). Seventeen patients underwent exome sequencing; their clinical characteristics are reported in Table S1. All included patients were TESE-negative, meaning that spermatozoa were not found in any of the multiple testis fragments.
Follow-up cohorts: exome data were available for 73 and 57 (total = 130) idiopathic MA subjects from the GEMINI consortium and the MERGE study, respectively. These samples were used only for targeted look-up of variants in the five novel NOA genes (See Supplementary Material and Methods).
Ethics statement
All patients (and family members where applicable) provided written informed consent to be included in the analyses and specifically exome sequencing. The study protocol was approved by the local ethics committees (Fundacio Puigvert: 2014/04c, Münster: 2010-578-f-S, GEMINI: 201502059). All experiments were performed in accordance with relevant guidelines and regulations.
Molecular genetics and statistical analyses
In the initial study, exome sequencing was carried out as a service by Macrogen Inc. (Republic of Korea) utilizing the Agilent SureSelect_V6 enrichment. Details on variant validation, phasing of STAG3 and SHOC1 variants and statistical analyses are provided in Supplementary Material and Methods.
Expression and meiotic studies in testis biopsy
Quantitative polymerase chain reaction (qPCR) was performed for ADAD2 expression analysis in testis biopsies from patient 11–151 and a collection of diverse histologies for comparison. Meiotic studies, based on fluorescent immunohistochemical staining, were performed in testicular tissue from the TERB1, SHOC1, RAD21L1, MSH4, MEIOB, SYCE1, and TEX11 variant carriers as described previously5,9 (See Supplementary Material and Methods).
RESULTS
In our discovery cohort of TESE-negative MA patients, we identified a plausible genetic cause in (1) 4/5 patients with incomplete SGA, (2) 0/2 patients with incomplete SCA, and (3) all 10 patients with complete SCA. Molecular and clinical data are summarized in Table 1. No other plausible genetic causes of NOA were identified among the filtered variants in these patients (Table S2).
Genetic defects in incomplete SGA: ADAD2, TEX14, and DMRT1
Novel NOA gene
-
Patient 11–151 carries a homozygous stopgain variant NM_139174.3:c.1186C>T; p.(Gln396Ter) in ADAD2. This may result in production of a truncated protein, whereby the adenosine–deaminase domain necessary for RNA binding and processing is affected. The fertile brother is a heterozygous carrier. qRT-PCR analysis of testicular RNA showed germ cell specific expression and a higher expression in SGA compared with SCA cases. ADAD2 expression was reduced approximately 2.7-fold for patient 11–151 compared with other SGA patients with intact ADAD2 (Fig. S1).
Known NOA genes
-
Patients 11–063 and 11–382 carry TEX14 loss of function (LoF) variants: 11–063 is compound heterozygous for a ~38-Kb deletion comprising exons 6 to 21 NM_001201457.1:c.(554+1_555-1)_(3378+1_3378-1)del p. (185del941aa) removing the protein kinase domain and a frameshift deletion NM_001201457.1:c.2303_2306del; p.(Gln768ArgfsTer31) in exon 14, generating a stopgain at amino acid (aa) 799 (total protein length: 1497 aa). Patient 11–382 carries a homozygous stopgain NM_001201457.1:c.3454C>T;p.(Arg1152Ter), which removes the last 345 aa.
-
Patient 17–204 carries a heterozygous deletion of exons 1 and 2 NM_021951.3:c.1_540del;p.(1del180aa) of DMRT1, resulting in removal of the first 180 aa of the protein, including the entire DM/DNA-binding domain. DNA of the parents was not available to define the deletion’s origin.
Genetic defects in complete SCA: TERB1, SHOC1, MSH4, RAD21L1, SYCE1, TEX11, MEIOB, MEI1, and STAG3
Novel NOA genes
-
Patient 10–200 carries two novel LoF variants in TERB1: a frameshift deletion NM_001136505.2:c.289_290del;p.(Leu97ValfsTer7), leading to a premature stop codon at aa 103, removing the entire SANT/Myb domain predicted to be involved in DNA-binding, and a stopgain at aa 605 NM_001136505.2:c.1813C>T;p.(Arg605Ter) (total protein length: 727 aa). The patient’s azoospermic brother carries the same genotype, and the heterozygosity for the frameshift deletion in the mother indicates compound heterozygosity in the two brothers (Fig. 1).
Fig. 1: Investigation of patient 10–200 carrying the TERB1 variant. 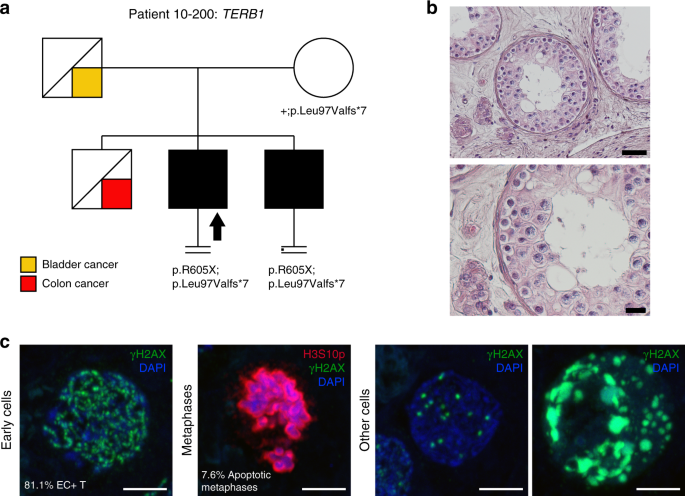
(a) The pedigree structure shows the segregation of the TERB1 variants (p.R605Ter and p.Leu97Valfs*7). (b) Hematoxylin and eosin (H&E) staining of histological sections from the testis biopsy of the patient carrying the TERB1 variant. Scale bar on the upper image represents 50 µm and on the lower image 20 µm. (c) Immunofluorescent staining of histological sections from the testis biopsy of the patient carrying the TERB1 variant using γH2AX (green, marker for DSBs and XY body), Histone 3 Serine 10 phosphorylation (H3S10p, red, marker of M-phase), and DAPI (blue). Scale bar represents 5 µm. In this patient only a few metaphases were observed (1.1 metaphases/mm2), which were below the normal threshold, based on samples containing meiotic metaphases (4.0–11.5 metaphases/mm2),9 thus representing the mitotic metaphases of spermatogonia. Early cells were observed in almost all the counted tubules. No XY bodies were detected, indicating that the cells fail to reach the pachytene stage. Most of the early cells displayed an aberrant pattern of γH2AX spots. Some cells displayed an aberrant pattern whereby γH2AX patches covered the entire nucleus.
-
Patient 11–272 carries a novel homozygous frameshift deletion NM_173521.4:c.797delT;p.(Leu266GlnfsTer6) in SHOC1 leading to a premature stop codon at aa 271 (total protein length: 1144 aa). The patient’s brother, who is also affected by complete SCA, carries the same genotype (Fig. 2).
Fig. 2: Investigation of patient 11–272 carrying the SHOC1 variant. 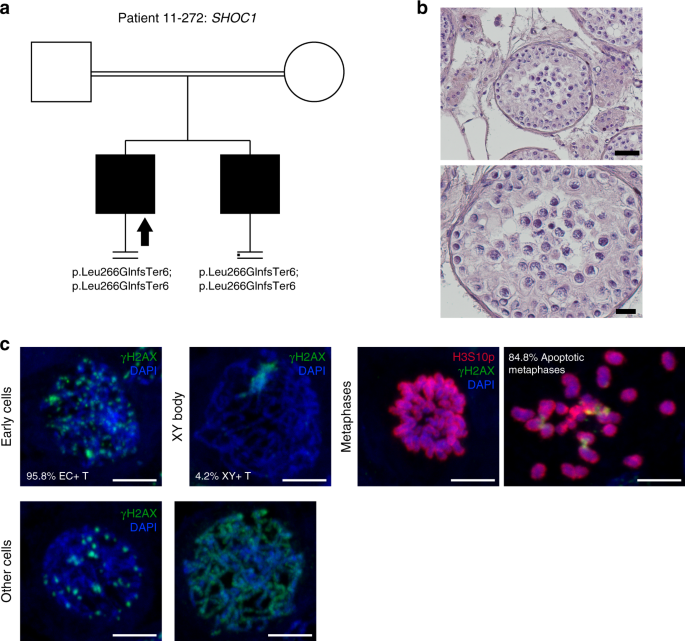
(a) The pedigree structure shows the segregation of the SHOC1 variant (p.Leu266GlnfsTer6). (b) Hematoxylin and eosin (H&E) staining of histological sections from the testis biopsy of the patient carrying the SHOC1 variant. Scale bar on the upper image represents 50 µm and on the lower image 20 µm. (c) Immunofluorescent staining of histological sections from the testis biopsy of the patient’s brother carrying the SHOC1 variant using γH2AX (Green), H3S10p (red), and DAPI (blue). Scale bar represents 5 µm. Early cells were observed in 95.8% of the round tubules that were counted. Extremely reduced XY body positive tubules were observed, indicating that the cells rarely reach the pachytene stage. A high density of metaphases was observed (29 metaphases/mm2) with a high percentage of apoptotic metaphases (84.8%). The organization of the apoptotic metaphases appeared to be more chaotic, as the chromosomes within these metaphases were dispersed. The early cells displayed an aberrant pattern of γH2AX spots. Some cells displayed an aberrant pattern whereby γH2AX is localized on the entire axis of the DNA strands.
-
Patient 11–127 carries a novel homozygous missense variant NM_002440.4:c.1913C>T;p.(Pro638Leu) in MSH4, affecting a highly conserved aa and predicted as pathogenic by all in silico tools (Fig. 3). The patient has two infertile sisters, but DNA from family members was not available for analysis.
Fig. 3: Investigation of patient 11–127 carrying the MSH4 variant. 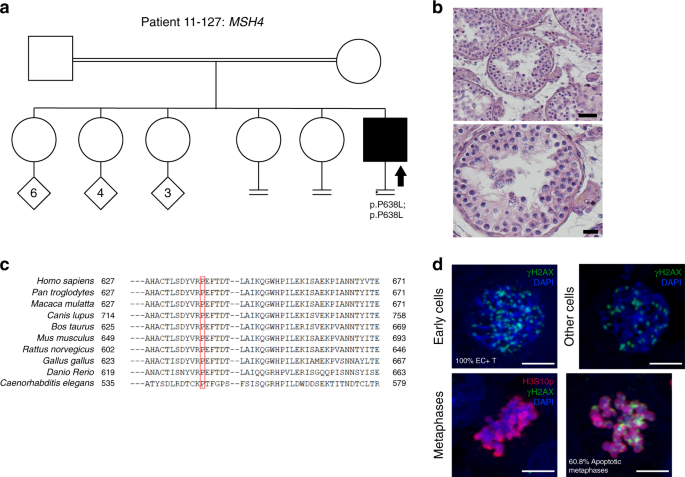
(a) The pedigree structure shows the segregation of the MSH4 variant (p.P638L). (b) Hematoxylin and eosin (H&E) staining of histological sections from the testis biopsy of the patient carrying the MSH4 variant. Scale bar on the upper image represents 50 µm and on the lower image 20 µm. (c) Sequence alignment of MSH4 protein orthologs, performed by HomoloGene. Red box highlights the Proline that is changed to Leucine in the MSH4 variant, which is highly conserved among species. (d) Immunofluorescent staining of histological sections from the testis biopsy of the patient carrying the MSH4 variant using γH2AX (green), H3S10p (red), and DAPI (blue). Scale bar represents 5 µm. Early cells were observed in 100% of the round tubules that were counted. In a striking number of early cells we observed a spotty γH2AX pattern. No XY bodies were observed in the tubules that were counted for this patient. The metaphase density is higher (15.2 metaphases/mm2) than the previously described control group. This indicates that there are meiotic metaphases present in this patient despite the lack of XY bodies. In addition, 60.8 % of the metaphases were positive for γH2AX.
-
Patient 07–359 carries a homozygous stopgain NM_001136566.2:c.1543C>T; p.(Arg515Ter) in RAD21L1, resulting in the removal of the last 41 aa of the protein (total length: 546 aa) eliminating the major part of a domain conserved with the other RAD21/REC8-like proteins. The fertile brother is a heterozygous carrier (Fig. 4).
Fig. 4: Investigation of patient 07–359 carrying the RAD21L1 variant. 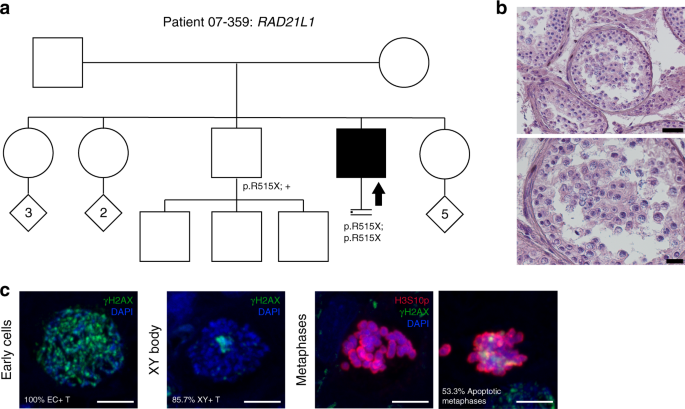
(a) The pedigree structure shows the segregation of the RAD21L1 variant (p.R515X). (b) Hematoxylin and eosin (H&E) staining of histological sections from the testis biopsy of the patient carrying the RAD21L1 variant. Scale bar on the upper image represents 50 µm and on the lower image 20 µm. (c) Immunofluorescent staining of histological sections from the testis biopsy of the patient carrying the SHOC1 variant using γH2AX (green), H3S10p (red), and DAPI (blue). Scale bar represents 5 µm. Early cells were observed in 100% of the round tubules that were counted. XY bodies were present in 85.5% of the tubules. The metaphase density is comparable with the control group (9.4 metaphases/mm2) however, a high percentage of these metaphases are apoptotic (53.3%).




Known NOA genes
-
Patient 15–285 carries a homozygous deletion of the entire open reading frame of the SYCE1 NM_001143764.3:c.1_1113del;p.(1del371aa) and CYP2E1 genes.
-
Patient 09–297 carries a hemizygous deletion of exons 4 to 9 in TEX11 NM_001003811.2:c.84_651del; p.(28del189aa). The deletion removes part of the meiosis specific protein Spo22/ZIP4/TEX11 domain.
-
Patients 08–079 and 09–167 carry MEIOB variants: 08–079 carries a novel homozygous deletion of the last 177 aa (4 exons) NM_001163560.3:c.897_1431del; p.(219del178aa). Patient 09–167 carries a homozygous frameshift deletion in exon 12 that is predicted to produce a premature stopgain at aa 381 NM_001163560.3:c.1140_1143del;p.(Asp381Ter) (total protein length: 471 aa). Both variants are predicted to affect the interaction with SPATA22.
-
Patient 18–406 is compound heterozygous for two likely pathogenic missense variants in exons 8 and 9 of MEI1 NM_152513.4:c.1088C>T;p.(Thr363Met) and NM_152513.4:c.925C>T;p.(Leu309Phe). Both mutated residues are conserved among species from rooster to primates (Figure S2), and the one in exon 8 is also present in the Arabidopsis thaliana MEI1 ortholog.10 The infertile sister carries both variants whereas the mother carries only one p.(Thr363Met).
-
Patient 13–567 carries two novel heterozygous LoF variants in STAG3: a splicing variant in the canonical position (+1) in intron 2, NC_000007.14:g.100180673del; and a frameshift deletion in exon 16, NM_012447.4:c.1645_1657del;p.(His549AlafsTer9). The frameshift deletion leads to a premature stop codon at aa 558 affecting the armadillo (ARM)-type domain. Long-range sequencing demonstrated that the variants are biallelic (compound heterozygous).
Meiotic analyses in SCA patients
Meiotic progression was assessed by immunofluorescent staining of specific markers to classify different types of arrest, and subsequent quantification of specific features (Table S3).5,9 Failure to form the XY body (silenced chromatin of the X and Y chromosomes) is a typical feature of many mouse meiotic mutants that display pachytene arrest, while meiotic metaphase arrest is marked by the presence of apoptotic metaphases.
For patients 10–200 (TERB1) and 15–285 (SYCE1), meiotic analyses revealed a pachytene arrest (no XY body formation; Fig. 1, Figures S3 and S4), with features of unrepaired meiotic DNA double-strand breaks (DSBs) in spermatocytes.
Spermatocytes of patients 11–272 (SHOC1), 11–127 (MSH4), and 09–297 (TEX11) also lacked XY bodies suggesting pachytene arrest, but still some spermatocytes apparently developed further, and the dramatic increase in the number of apoptotic metaphases indicated complete metaphase arrest (Figs. 2 and 3, Figures S5, S6, and S7).
Patient 07–359 (RAD21L1) and patients 08–079 and 09–167 (both MEIOB) displayed reduced XY body formation in combination with complete metaphase arrest, which was more similar to results obtained in a recently analyzed patient group9 (Fig. 4, Figures S8, S9, and S10).
Replication study of the novel causes of MA
The five novel NOA genes were screened in 130 MA patients for whom exome data were available in the GEMINI and MERGE studies. For 4/5 genes, rare and predicted pathogenic variants were identified as the most plausible cause of MA in five unrelated patients. We observed a significantly higher frequency of homozygous LoF variants (in ADAD2, RAD21L1, and SHOC1) in the 147 patients (17 discovery and 130 validation), compared with that in the entire gnomAD exome database (p values ranging from 2 × 10−3 to 1 × 10−6) (Table S4). Moreover, we calculated the PopScore for each variant in the five new genes by using the population sampling probability pipeline (PSAP) and they were very low for all variants (ranging from 3.2 × 10−4 to 2.1 × 10−7) (for details see Supplementary information; Table S5).
Patient GEMINI-1020 is affected by incomplete SGA and is compound heterozygous for a ~212-kb deletion that includes ADAD2 (Hg19: chr16:84012049-84224913del) and a frameshift insertion NM_001145400.2:c.82dupC;p.(Gln28ProfsTer136) in exon 1 introducing a premature stop codon in the same gene at aa 136 (total protein length: 583 aa).
Patient M468 affected by SCA carries a rare homozygous missense and predicted as pathogenic variant NM_001136505.2:c.236C>T;p.(Ala79Val) in TERB1. The mutated aa is highly conserved among species from worm to primates.
Two unrelated MA patients carried recessive LoF variants in SHOC1. Patient M2012 is homozygous for a frameshift deletion NM_173521.4: c.1085_1086del;p.(Glu362ValfsTer25) and patient M2046 is compound heterozygous for NM_173521.4:c.945_948del; p.(Glu315AspfsTer6), NM_173521.4:c.1351del;p.(Ser451LeufsTer23) and NM_173521.4:c.1347T>A;p.(Cys449Ter). All variants are predicted to introduce a premature stopgain, which would remove 65–70% of the protein.
Patient M1916 affected by MA carries a rare homozygous missense variant NM_002440.4:c.2261C>T;p.(Ser754Leu) in MSH4 predicted as pathogenic by all in silico tools. The mutated aa is highly conserved among species from mosquito to primates.
DISCUSSION
Given the multifactorial and polygenic nature of NOA and the variety of testis phenotypes, here we applied exome sequencing to a highly selected group of patients affected by idiopathic MA with spermatozoa neither histologically identified nor recovered through TESE by multiple testicular biopsies. We focused on this specific testis phenotype because the identification of genetic factors associated with negative TESE not only has diagnostic value but also prognostic value for TESE outcome. In our discovery cohort, we successfully identified ADAD2 as a novel cause of SGA, and four novel genes for SCA: TERB1, SHOC1, MSH4, and RAD21L1. There are several lines of evidence to support their relevance: (1) the mouse KO phenotype of these genes recapitulates the human phenotype; (2) segregation of the genotype with the phenotype among brothers was observed for TERB1, SHOC1, and heterozygous brothers of ADAD2 and RAD21L1 variants were fertile; (3) variants predicted as pathogenic were subsequently identified for ADAD2, TERB1, SHOC1, and MSH4 in independent validation cohorts of MA patients, and the observed testis phenotypes of the carriers matched the phenotypes of carriers in the discovery cohort; (4) our meiotic studies provided mechanistic explanation to the genotype/phenotype correlation; (5) the relevance of the variants in the five genes is supported by the very low PopScore; and (6) the frequency of homozygous LoF variants in our cohort versus gnomAD individuals was significantly higher. However, evidence from independent patients/pedigrees (and/or genome sequencing) would be necessary to definitively prove the causality of identified variants/genes for MA.
Apart from the five novel candidate genes, we validated seven genes (TEX14, DMRT1, SYCE1, TEX11, MEIOB, MEI1, and STAG3) that were previously reported in NOA individuals (Table S6). With the exception of DMRT1, recessive inheritance was observed for all genes. Interestingly, not all affected men were from consanguineous families and, thus, these genetic causes for NOA should also be considered in outbred populations. In the current literature the number of genes reported to be involved in nonsyndromic NOA is 29, and among them only 13 MA candidate genes were reported in more than one study. A small minority of them reached to sufficient clinical evidence for diagnostic testing according to Oud et al.3 Our study represents a major advancement, since for three genes TEX11, TEX14, and STAG3 there is now definitive/strong clinical evidence while clinical evidence for two genes, MEI1 and MEIOB, is upgraded to moderate (Table S6). Prior to this study, there was sufficient clinical evidence for six genes (DMRT1, HSF2, SYCP3, TEX11, TEX15, and XRCC2) to recommend diagnostic testing.3 The current study increases this number to ten, and those genes which are associated with complete MA are candidates for a future pre-TESE prognostic gene panel.
We propose five novel genetic etiologies for human MA and all are strong candidates since the phenotype of respective KO mice is similar. Of these, only one was associated with incomplete spermatogonial arrest in two patients who carried ADAD2 variants. ADAD2 belongs to the double-stranded RNA binding proteins (dsRBP); it binds with high affinity to highly structured RNA substrates, such as small noncoding RNAs (sncRNAs) where it may edit the RNA through its deaminase activity and thereby control RNA structure and/or protein coding sequence.11 According to the Mouse Genome Informatics database (http://www.informatics.jax.org/) the KO mouse displays male and female infertility. Based on the testis phenotypes of the patients from the two different cohorts and the observed reduced ADAD2 messenger RNA (mRNA) expression, we propose a critical role for ADAD2 in spermatogonia where it is likely acting as a dsRNA-binding protein.12 Concerning the other four new genes, different types of meiotic arrest were observed. First, in the TERB1 variant carriers, we observed a pachytene arrest, which recapitulates the mouse phenotype13 (Table S7). TERB1 is a testis specific telomere-associated protein, which is essential in the regulation of chromosome movement to promote homologous pairing during meiotic prophase I.13 Defects in the other three newly identified MA associated genes (SHOC1, RAD21L1, and MSH4) show metaphase arrest that was somewhat less severe compared with the mutant mouse models (Table S5). For SHOC1 we observed an almost complete lack of XY bodies (indicative of failure to complete chromosome pairing) but still progression to metaphase. SHOC1 is a homolog of the budding yeast Zip2 protein, which forms a tight complex with Zip4, homologous to mammalian TEX11.14 They interact with components of the synaptonemal complex and play a role in DSB repair and crossover formation. Shoc1 knockout in mice also affects XY body formation, but subsequently leads to a pachytene arrest.15 This may indicate that the variants allow production of a partially functional (truncated) protein, but we may also hypothesize possible differences in the stringency of meiotic arrest inducing checkpoints between mouse and human (see below). The second novel mutated gene associated with metaphase arrest, RAD21L1, is a testis specific component of the cohesion complex involved in meiotic chromosome pairing and separation.16 Rad21l KO male mice are defective in full synapsis (physical protein linkage between chromosomes) of homologous chromosomes at meiotic prophase I, provoking an arrest at a zygotene-like stage.16 In our RAD21L1 patient carrying the variant, XY body formation, indicating completion of synapsis, was observed in >70% of the tubules. Thus, the 41 C-terminal amino acids of this protein that are lacking in our patient may be essential for progression beyond meiotic metaphase, but possibly not for homologous chromosome synapsis. Finally, MSH4 encodes a member of the DNA mismatch repair mutS family, which is highly expressed in testis. It is required for reciprocal recombination and proper chromosome pairing during meiosis I. For MSH4, the variant in the ATPase domain analyzed in this study yields a less severe phenotype compared with that observed in the Msh4 mouse knockout,17 but is very reminiscent of the mouse Msh5 ATPase mutant phenotype.18 In this Msh5 point mutant, XY body formation was severely affected, but a subset of cells progressed to meiotic metaphase, indicating that some function of this heterodimer does not require ATPase activity, since the full knockout displays a more severe phenotype.19 Additionally, in this case, we observe a complete lack of XY body formation, but still an arrest at meiotic metaphase (further discussed below). In addition to these five novel genes associated with NOA, our detailed clinical description and meiotic studies also provide novel data for the genotype/phenotype correlation of seven previously reported genes. For two of them, TEX14 and DMRT1, the previously described testis phenotypes are heterogeneous (See Table S6). We identified two homozygous LoF variants in TEX14 in two unrelated patients affected by incomplete spermatogonial arrest. Previously reported TEX14 variants were associated with testis histology ranging from SCO to complete SCA.20,21,22 With the discovery of the two new TEX14 variant carriers, this gene can now be upgraded to strong clinical evidence. We conclude that TEX14 defects are a relatively frequent cause of NOA due to SCO or early MA with TESE-negative outcome. Another patient with early MA (incomplete SGA), carried a heterozygous deletion of exons 1 and 2 in DMRT1, for which deletions as well as rare nucleotide variants have been associated with different NOA phenotypes.23,24 The current study further increases the evidence for a pathogenic effect of DMRT1 haploinsufficiency.
Regarding the novel variants in TEX11, STAG3, MEIOB, MEI1, and SYCE1, our findings are consistent with earlier observations, and confirm the critical role of these genes in meiosis, i.e., all our patient showed complete SCA (Table 1). Most importantly, thanks to our study, TEX11 has reached the level of definitive clinical evidence. For most of these patients, we were able to obtain novel information about the exact nature of meiotic arrest. MEIOB is a single-stranded DNA (ssDNA) binding protein and a meiosis-specific paralog of the more well-known repair protein RPA1. MEIOB functions together with another meiosis-specific protein, SPATA22, in the processing of early meiotic recombination intermediates. Knockout of either Meiob25 or Spata2226 in mouse results in very severe defects in meiotic DSB repair, resulting in what is generally called pachytene arrest, whereby cells have a more zygotene-like appearance, because homologous chromosomes fail to pair and synapse. Based on the shared region of MEIOB in patients 09–167 and 08–079 that is still intact, it seems most likely that the 293 N-terminal amino acids allow the truncated MEIOB to aid in completion of DSB repair for at least a portion of the meiotic DSBs, resulting in complete chromosome pairing in at least part of the cells as evidenced by the (reduced) XY body formation observed. However, crossover formation is severely affected, resulting in an arrest at metaphase I instead of at pachytene. It is noteworthy that the MEIOB variants observed in our study and in previous studies cluster in exon 12, suggesting a hotspot variant region, at least in the Arab/Pakistani population.27,28 For SYCE1 we observed a pachytene arrest (XY body formation failure), consistent with phenotypes in mice. For our TEX11 patient, the complete lack of XY bodies was associated with arrest occurring at metaphase instead of at pachytene. This unexpected phenotype is very similar to phenotypes we observed for the SHOC1 and MSH4 variant carriers. Pachytene arrest in mice is very robust,29 and observed in 7/9 of the knockout mice for the meiotic genes found mutated in this paper (Table S7). It is triggered by a DNA damage response, and by incomplete meiotic sex chromosome inactivation (MSCI, failure to form the XY body).29 Therefore, in the patients carrying variants in MSH4, TEX11, or SHOC1, the (near) complete absence of cells with XY bodies would be expected to trigger such a pachytene arrest, but we observed a later, metaphase arrest. However, previous analyses have indicated that MSCI is more variable in human than mouse, or perhaps not even achieved in all cells that complete meiosis.30,31 Together with our observations, this could indicate that a failure of XY body formation in humans may not always lead to a pachytene arrest. This may also explain the overall more frequent occurrence of metaphase arrest compared with pachytene arrest in human compared with mouse, as a cause of spermatocyte arrest.9
An emerging issue in the field of human reproduction is that some genetic factors may affect both male and female fertility. In males, meiotic defects typically result in NOA, whereas in females they are associated with premature ovarian insufficiency (POI). We report variants in MSH4, SYCE1, MEIOB, and MEI1 in males with complete SCA. Variants in these genes were also previously reported to cause POI or hydatidiform mole in females.28,32,33,34 Interestingly, both of our patients with variants in MSH4 and SYCE1 had at least one infertile sister. Although it is not known whether our novel MA genes ADAD2, RAD21L1, and TERB1 are involved in POI, knockout of these genes in mice results in infertility in both sexes.13,17,35 Hence, we encourage further research on shared genetic factors causing both male and female infertility. The identification of such genetic defects implies that genetic counseling for NOA has relevance to both male and female family members, especially because in the latter, fertility preservation could be advised prior to ovarian failure.
Our study also has wider implications. First, the discovery of novel genetic factors and their introduction to a diagnostic setting opens novel avenues for their potential relevance to the general health status of NOA men. It is clear that spermatogenesis and tumorigenesis may share common genetic factors, and according to a growing body of evidence, reproductive phenotype may serve as a biomarker of general health.36,37 NOA is a multifactorial condition and the identification of the underlying genetic defect through diagnostic genetic testing would allow the identification of patients at higher risk for general health problems. Recently, starting from exome analysis performed with the purpose of diagnosing the etiology of SCO, we identified three subjects with “occult”, late onset Fanconi anemia. These patients are now under surveillance with oncohematologists.38 In our current paper, three of the five new MA genes (TERB1, MSH4, and RAD21L1) are involved in the maintenance of genome integrity and two patients are variant carriers of another DNA repair gene, MEIOB. Although these genes are not considered among the 32 cancer predisposition genes39 their defective function may increase risk for the acquisition of variants in cancer genes leading to cancer predisposition.40 Second, the genetic dissection of meiotic arrest through genotype/phenotype correlation is also of relevance for the development of male contraceptives based on germ cell specific targeting.
In conclusion, we describe a highly successful genetic screening contributing substantially to the development of a pre–testis biopsy gene panel. If properly validated, the genetic diagnosis of complete MA prior to surgical interventions, aimed at the recovery of testicular spermatozoa for subsequent in vitro fertilization, will potentially prevent such procedures if the chances of success are very low or virtually zero. Hence, the identification and validation of gene defects associated with poor TESE prognosis is critical in informing surgical decisions in about 1% of men in the general population.
Change history
07 August 2020
The original online PDF version of the Article contained figures in monochrome. They now appear in colour in the PDF and HTML versions of the Article.
References
Krausz C, Riera-Escamilla A. Genetics of male infertility. Nat Rev Urol. 2018;15:369–384.
Tournaye H, Krausz C, Oates RD. Novel concepts in the aetiology of male reproductive impairment. Lancet Diabetes Endocrinol. 2017;5:544–553.
Oud MS, Volozonoka L, Smits RM, Vissers LELM, Ramos L, Veltman JA. A systematic review and standardized clinical validity assessment of male infertility genes. Hum Reprod. 2019;34:932–941.
van der Bijl N, Ropke A, Biswas U, et al. Mutations in the stromal antigen 3 (STAG3) gene cause male infertility due to meiotic arrest. Hum Reprod. 2019;34:2112–2119.
Riera-Escamilla A, Enguita-Marruedo A, Moreno-Mendoza D, et al. Sequencing of a “mouse azoospermia” gene panel in azoospermic men: identification of RNF212 and STAG3 mutations as novel genetic causes of meiotic arrest. Hum Reprod. 2019;34:978–988.
Schilit SLP, Menon S, Friedrich C, et al. SYCP2 translocation-mediated dysregulation and frameshift variants cause human male infertility. Am J Hum Genet. 2020;106:41–57.
Yatsenko AN, Georgiadis AP, Ropke A, et al. X-linked TEX11 mutations, meiotic arrest, and azoospermia in infertile men. N Engl J Med. 2015;372:2097–2107.
Krausz C, Hoefsloot L, Simoni M, Tuttelmann F. EAA/EMQN best practice guidelines for molecular diagnosis of Y-chromosomal microdeletions: state-of-the-art 2013. Andrology. 2014;2:5–19.
Enguita-Marruedo A, Sleddens-Linkels E, Ooms M, et al. Meiotic arrest occurs most frequently at metaphase and is often incomplete in azoospermic men. Fertil Steril. 2019;112:1059–1070.e3.
De Muyt A, Vezon D, Gendrot G, Gallois J-L, Stevens R, Grelon M. AtPRD1 is required for meiotic double strand break formation in Arabidopsis thaliana. EMBO J. 2007;26:4126–4137.
de Mateo S, Sassone-Corsi P. Regulation of spermatogenesis by small non-coding RNAs: role of the germ granule. Semin Cell Dev Biol. 2014;29:84–92.
Wang X, Vukovic L, Koh HR, Schulten K, Myong S. Dynamic profiling of double-stranded RNA binding proteins. Nucleic Acids Res. 2015;43:7566–7576.
Wang Y, Chen Y, Chen J, et al. The meiotic TERB1-TERB2-MAJIN complex tethers telomeres to the nuclear envelope. Nat Commun. 2019;10:564.
Lynn A, Soucek R, Borner GV. ZMM proteins during meiosis: crossover artists at work. Chromosome Res. 2007;15:591–605.
Zhang Q, Shao J, Fan H-Y, Yu C. Evolutionarily-conserved MZIP2 is essential for crossover formation in mammalian meiosis. Commun Biol. 2018;1:147.
Herran Y, Gutierrez-Caballero C, Sanchez-Martin M, et al. The cohesin subunit RAD21L functions in meiotic synapsis and exhibits sexual dimorphism in fertility. EMBO J. 2011;30:3091–3105.
Kneitz B, Cohen PE, Avdievich E, et al. MutS homolog 4 localization to meiotic chromosomes is required for chromosome pairing during meiosis in male and female mice. Genes Dev. 2000;14:1085–1097.
Milano CR, Holloway JK, Zhang Y, et al. Mutation of the ATPase domain of MutS homolog-5 (MSH5) reveals a requirement for a functional MutSgamma complex for all crossovers in mammalian meiosis. G3 (Bethesda). 2019;9:1839–1850.
de Vries SS, Baart EB, Dekker M, et al. Mouse MutS-like protein Msh5 is required for proper chromosome synapsis in male and female meiosis. Genes Dev. 1999;13:523–531.
Gershoni M, Hauser R, Yogev L, et al. A familial study of azoospermic men identifies three novel causative mutations in three new human azoospermia genes. Genet Med. 2017;19:998–1006.
Fakhro KA, Elbardisi H, Arafa M, et al. Point-of-care whole-exome sequencing of idiopathic male infertility. Genet Med. 2018;20:1365–1373.
Fenz Araujo T, Friedrich C, Paiva Grangeiro CH, et al. Sequence analysis of 37 candidate genes for male infertility: challenges in variant assessment and validating genes. Andrology. 2020;8:434–441.
Lopes AM, Aston KI, Thompson E, et al. Human spermatogenic failure purges deleterious mutation load from the autosomes and both sex chromosomes, including the gene DMRT1. PLoS Genet. 2013;9:e1003349.
Tewes A-C, Ledig S, Tuttelmann F, Kliesch S, Wieacker P. DMRT1 mutations are rarely associated with male infertility. Fertil Steril. 2014;102:816–820.
Souquet B, Abby E, Herve R, et al. MEIOB targets single-strand DNA and is necessary for meiotic recombination. PLoS Genet. 2013;9:e1003784.
Hays E, Majchrzak N, Daniel V, et al. Spermatogenesis associated 22 is required for DNA repair and synapsis of homologous chromosomes in mouse germ cells. Andrology. 2017;5:299–312.
Gershoni M, Hauser R, Barda S, et al. A new MEIOB mutation is a recurrent cause for azoospermia and testicular meiotic arrest. Hum Reprod. 2019;34:666–671.
Caburet S, Todeschini A-L, Petrillo C, et al. A truncating MEIOB mutation responsible for familial primary ovarian insufficiency abolishes its interaction with its partner SPATA22 and their recruitment to DNA double-strand breaks. EBioMedicine. 2019;42:524–531.
Royo H, Polikiewicz G, Mahadevaiah SK, et al. Evidence that meiotic sex chromosome inactivation is essential for male fertility. Curr Biol. 2010;20:2117–2123.
de Vries M, Vosters S, Merkx G, et al. Human male meiotic sex chromosome inactivation. PLoS ONE. 2012;7:e31485.
Mulugeta Achame E, Baarends WM, Gribnau J, Grootegoed JA. Evaluating the relationship between spermatogenic silencing of the X chromosome and evolution of the Y chromosome in chimpanzee and human. PLoS ONE. 2010;5:e15598.
Carlosama C, Elzaiat M, Patino LC, Mateus HE, Veitia RA, Laissue P. A homozygous donor splice-site mutation in the meiotic gene MSH4 causes primary ovarian insufficiency. Hum Mol Genet. 2017;26:3161–3166.
de Vries L, Behar DM, Smirin-Yosef P, Lagovsky I, Tzur S, Basel-Vanagaite L. Exome sequencing reveals SYCE1 mutation associated with autosomal recessive primary ovarian insufficiency. J Clin Endocrinol Metab. 2014;99:E2129–E2132.
Nguyen NMP, Ge Z-J, Reddy R, et al. Causative mutations and mechanism of androgenetic hydatidiform moles. Am J Hum Genet. 2018;103:740–751.
Dickinson ME, Flenniken AM, Ji X, et al. High-throughput discovery of novel developmental phenotypes. Nature. 2016;537:508–514.
Eisenberg ML, Li S, Brooks JD, Cullen MR, Baker LC. Increased risk of cancer in infertile men: analysis of U.S. claims data. J Urol. 2015;193:1596–1601.
Nagirnaja L, Aston KI, Conrad DF. Genetic intersection of male infertility and cancer. Fertil Steril. 2018;109:20–26.
Krausz C, Riera-Escamilla A, Chianese C, et al. From exome analysis in idiopathic azoospermia to the identification of a high-risk subgroup for occult Fanconi anemia. Genet Med. 2019;21:189–194.
LaDuca H, Polley EC, Yussuf A, et al. A clinical guide to hereditary cancer panel testing: evaluation of gene-specific cancer associations and sensitivity of genetic testing criteria in a cohort of 165,000 high-risk patients. Genet Med. 2020;22:407–415.
Kim Y-H, Ohta T, Oh JE, et al. TP53, MSH4, and LATS1 germline mutations in a family with clustering of nervous system tumors. Am J Pathol. 2014;184:2374–2381.
Acknowledgements
The authors thank the patients participating in the study for their important contribution. A special acknowledgment is dedicated to Esperança Martí (President of the Fundació Puigvert). Esther Sleddens-Linkels, Jochen Wistuba, and Jutta Salzig are gratefully acknowledged for their help with testicular histological evaluation. We thank Christian Ruckert, Jochen Seggewiß, and Marius Wöste for their support in bioinformatics and the long-range seq, respectively. The study was supported by Spanish Ministry of Health Instituto Carlos III-FIS (grant number FIS/FEDER-PI14/01250; PI17/01822to C.K. and A.R.-E.), the European Commission, Reproductive Biology Early Research Training (REPROTRAIN, project number 289880, awarded to C.K. and A.R.-E.), the German Research Foundation Clinical Research Unit “Male Germ Cells: from Genes to Function” (DFG CRU326, grants to F.T.), and by the National Institutes of Health (R01HD078641, awarded to D.F.C., L.N., K.I.A., and D.T.C.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Krausz, C., Riera-Escamilla, A., Moreno-Mendoza, D. et al. Genetic dissection of spermatogenic arrest through exome analysis: clinical implications for the management of azoospermic men. Genet Med 22, 1956–1966 (2020). https://doi.org/10.1038/s41436-020-0907-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0907-1
Key words
This article is cited by
-
A novel homozygote nonsense variant of MSH4 leads to primary ovarian insufficiency and non-obstructive azoospermia
Molecular Biology Reports (2024)
-
A report of two homozygous TERB1 protein-truncating variants in two unrelated women with primary infertility
Journal of Assisted Reproduction and Genetics (2024)
-
Novel MEI1 mutations cause chromosomal and DNA methylation abnormalities leading to embryonic arrest and implantation failure
Molecular Genetics and Genomics (2024)
-
Frequency, morbidity and equity — the case for increased research on male fertility
Nature Reviews Urology (2024)
-
Identification of compound heterozygous variants in MSH4 as a novel genetic cause of diminished ovarian reserve
Reproductive Biology and Endocrinology (2023)
