
Abstract
Purpose
The Electronic Medical Records and Genomics (eMERGE) Consortium integrated biorepository-based research with electronic health records (EHR) to return results from large-scale genetic tests to participants and uploaded those data into the EHR. This article explores the ethical issues investigators encountered in that process.
Methods
We conducted in-depth, semistructured interviews with study personnel of the eMERGE-III Consortium sites that returned results.
Results
We discuss major ethical issues that arose while attempting to return research results from the eMERGE Consortium to individual participants. These included difficulties recontacting those participants who had not explicitly consented to such and disclosing results to many participants with insufficient infrastructure and staff. Investigators reported being driven by a supererogatory clinical impulse.
Conclusion
All these issues ultimately derive from ethical conflicts inherent to translational work being done at the interface of research and clinical care. A critical rethinking of this divide is important, but infrastructural support for such work is necessary for an ethically sound rollout of large-scale genetic testing.
Similar content being viewed by others


INTRODUCTION
The practice of medicine in the United States has historically made ethical and legal distinctions between therapy and research.1 This has been referred to as the segregation model.2 The two functions have always occurred in conjunction, but they have been treated analytically as relatively exclusive domains. Yet maintaining the boundary between research and clinical activities was “the most difficult and complex problem” for the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research.2 Indeed, studies have shown that patients have difficulty differentiating the two domains.1,3 Even institutional review board personnel struggle in this regard.4 A major reason for this confusion is what Wolf and colleagues call the “interdigitation” of research and therapy.5 Many forms of clinical practice regularly engage in research as a mode of intervention, such as clinical trials for cancer patients.6 Moreover, the two activities often take place in academic medical centers. Patients may be identified or recruited to participate by their clinicians who might themselves simultaneously serve as the investigators. Research candidates might even be enticed into participation by the “therapeutic benefit” of the individual results promised to be returned to them.7,8
The conflation of research and clinical care is particularly prevalent and potentially problematic in translational research, which seeks to understand whether implementing the products of basic research in clinical settings improves health outcomes, and if so, how best to proceed. This process is often referred to as T2 research.9
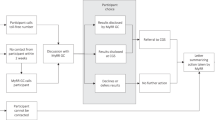
The Electronic Medical Records and Genomics (eMERGE) Consortium provides a particularly illuminating version of this kind of boundary blurring between clinical care and research as well as the ethical dilemmas for investigators that result from that blurring. The eMERGE Consortium has involved networks of medical institutions across the United States that integrate the results of genomic research with electronic health records (EHRs) for discovery. As a part of the third phase of the study, nine sites returned genetic research results to some or all of their participants to assess the impact of these results on clinicians and participants. Each site developed its own process for recruiting participants—most of whom had no clinical indication for testing—and returning results of variants in up to 109 genes, details of which have been described elsewhere.10 Notably, some sites obtained consent to return results during recruitment, but others, which typically had recruited participants previously when they had not anticipated returning results, sought consent to return only after concerning results had been received. In eMERGE-III, therefore, returning results to individual participants and their providers is a major part of the research itself, making it an instructive example of work that transcends the research/clinical care dichotomy.11
In this article, we report the ethical quandaries encountered by individuals intimately involved in designing and implementing the return of results in eMERGE sites.10 We argue that the very process of translational research poses moral dilemmas, in part because the interventions are not yet accepted parts of the clinical practice that the research seeks to explore and the pathways for delivery are not yet established. In particular, we contend that—especially for clinicians serving in a separate role as research investigator, even if not vis-à-vis their own patients—walking the line between research and clinical care can result in a form of moral distress that we call the clinical impulse. This impulse obliged researchers to treat eMERGE results as though they had been obtained as a part of accepted clinical practice. Knowledge of potentially medically relevant information about participants led many sites to undertake difficult measures, often involving research study workarounds, to provide information and follow-up care to participants. We discuss the ethical implications of the clinical impulse below.
MATERIALS AND METHODS
Study personnel (SP) of the eMERGE-III Consortium sites who were involved in designing and implementing the return of results process were sent an initial email, inviting them to participate in an hour-long interview discussing ethical issues related to the return of results through eMERGE-III. An in-depth, semistructured interview guide was developed by our research team after reviewing existing literature and in consultation with other methodologists and skilled qualitative researchers at Vanderbilt University Medical Center. The guide addressed each site’s unique procedure for recruiting, consenting, and returning potentially pathogenic genetic variants. We also explored unexpected difficulties in recontacting and returning results to participants; unexpected difficulties in interpreting results, which was done centrally with further work at many sites; responses of SP involved in returning the results; and responses of participants involved in the eMERGE study. The interview was pretested with four individual members of the eMERGE Consortium from four different sites (Cincinnati Children’s Hospital Medical Center, Geisinger Health System, and Northwestern University).12 This study focused only on challenges raised by addressing potentially pathogenic variants.
Data analysis
All interviews were conducted by the first author (C.M.E.H.), who is an experienced interviewer trained in qualitative methods. After receiving informed consent, they were audio-recorded and transcribed verbatim. Transcripts were uploaded into Dedoose—a web-based, mixed-methods analysis tool—where they were coded by two researchers (C.M.E.H., E.W.C.). A coding tree was developed jointly and iteratively, based on discussions of themes that emerged from the interviews and using conventional content analysis.13 All coding conflicts were readily resolved through discussion to reach consensus, and intercoder reliability (>80%) was achieved.14 We focused the present analysis on ethical quandaries faced by investigators related to the return of results in such translational settings.
Ethics statement
This study was approved by the Vanderbilt University Medical Center IRB, and all participants gave verbal consent for their participation.
RESULTS
Response rate and demographics
Study personnel who participated in the interview came from each of the nine sites that had returned results: Cincinnati Children’s Hospital Medical Center (CCHMC), Children’s Hospital of Philadelphia (CHOP), Columbia University (CU), Geisinger Health System (GHS), Harvard University (HU), Kaiser Permanente/University of Washington (KP/UW), Mayo Clinic (MC), Northwestern University (NU), and Vanderbilt University Medical Center (VUMC). Twenty-seven individuals were invited to complete an interview. A total of 20 individuals (74%) participated in our study, 14 of whom also work as clinicians (of whom 8 are genetic counselors), while the remainder comprised project managers and social scientists. This includes the four cognitive interviews we initially conducted. Interviews lasted an average of 57 minutes and took place between April and May 2019.
Several ethical concerns arose consistently during the interviews, all resulting from the translational nature of the study. These concerns spanned the entire scope of the return of results process: from initial attempts to recontact participants—first, to obtain consent to return results for those whose initial consent did not permit this, and then, once results were available, to disclose clinically actionable results—to issues arising from the scale of results disclosure, and finally to the question of how to finance such efforts. Each of these issues generated a form of moral distress that led eMERGE personnel to strive to overcome perceived barriers imposed by the research study to provide participants with what amounted to clinical care.
Recontacting participants for enrollment
Several sites (CCHMC, CHOP, HU, KP/UW, GHS, NU) decided to recontact biobank participants after results had been obtained to get specific consent to return study results that personnel determined to be of clinical relevance. Some consent forms previously signed by biobank participants did not allow for the return of results. Some biobanks even promised participants that they would never be recontacted. Some interviewees specifically worried that it would be inappropriate to heed participants’ refusal to get results. “The question is always: How well did they really understand the consent process when you have this actionable variant? Because ethically, you have got to really understand what they’re declining” (KP/UW). These interviewees believed that it was unclear that participants in such a biobank could have sufficiently understood what they were giving up. They were skeptical that enrollees would decline disclosure if they had truly understood the potential impact of receiving results.
Some investigators felt acutely that returning results seemed not merely warranted but morally required. Yet investigators worried that simply returning results without obtaining prior consent would have been unethical. Therefore, some sites decided to approach the recontact for revised consent to permit returning research results indirectly: “We had to tell participants, ‘Look, we said we would never reestablish the data link, but if we had the chance, can we do it?’” (CHOP). Without revealing that testing had taken place or any specifics of the results, participants who had initially enrolled without any presumption of personal benefit were asked to consent again to permit such disclosure. At least anecdotally, interviewees reported that no participant had complained that they felt the recontact to be a breach of their initial consent; rather, they incorrectly believed that their initial consent guaranteed that they would receive any relevant results.
Researchers obtained new consent despite the significant burdens recontact incurred in terms of both time and resources. “I think it was very difficult, definitely an added difficulty” (CHOP), said one project manager, recounting the added work hours and other resources required to consent participants in this way. Locating individuals who had enrolled in a given biobank years prior was often difficult, since with the lapse of time, available contact information was often invalid. The process of obtaining new consent significantly slowed the eMERGE-III timeline for these sites. But in the end, personnel often believed it was worth the burdens it imposed: “I’m sitting on big pathogenic results” (KP/UW), one clinician worried, but she was not permitted to return them until specific consent was obtained from the affected participants.
Many interviewees also discussed the difficulty of returning results to family members of deceased participants (CU, GHS, KP/UW, MC, NU, HU, VUMC) and to patients who had been enrolled as minors but had subsequently reached legal majority (CCHMC, CHOP). Interviewees characterized some of these issues as hurdles that might have been anticipated but simply had not been. For many of these sites, recontacting participants to return results in this fashion was even more complicated.
Scaling research
Many sites understood eMERGE to be a sort of incubator study, as an attempt to understand how best to bring genomic science to the bedside. However, interviewees acknowledged that asymmetries between research and clinical endeavors made it quite difficult to scale something intended to be clinically practicable into a nationwide study involving thousands of participants.
A task that can be challenging enough in the clinical setting—for example, returning potentially pathogenic genetic test results—can be extremely burdensome at a larger scale, due to a “scarcity of genetic counselors” (MC) and inadequate staff and infrastructure. Even the traditional means of recontact and return of results were stressed when dealing with so many participants. For instance, one site cautioned that “in the future, there [need to be] more reliable mechanisms than US postal mail when you’re working at such a large scale” (MC).
This ultimately led many sites to develop strategies that differed from routine clinical care. One genetic counselor opined, “In a diagnostic setting, most positive results are still going to be called out individually. In a population-screening setting, that’s where you’re going to need a better triage method for who needs a more extensive counseling session” (NU). As another clinician explained, the challenge in scaling this type of translational research was to decide “what patients would benefit from a genetic counseling session versus what patients are okay learning the information just in a letter or a video or something like that […] as opposed to having to call every single positive person” (NU). These issues were even more challenging for sites that sought to return all results, not just those that were potentially pathogenic. Returning results that did not include likely/pathogenic variants, which was the case for up to 95% of participants, increased the volume of work enormously, making this sort of scaling even more difficult.
Role mixing and moral distress
Many participants in our interviews either explicitly noted the blurring of boundaries in eMERGE between clinical care and research, or conflated the two in our conversations without noting that they were doing so. A genetic counselor told us that eMERGE “feels very clinical even though they’re research participants” (CU). A project manager at a different site said that even though “the [investigators] who created these protocols really thought about [eMERGE] as just a research project,” in practice these personnel were “essentially creating a temporary clinic where these patients have clinical results and you have to take care of them” (VUMC). In the same vein, a project manager described eMERGE-III as “a research encounter with clinically valid results” (MC).
The mixing of different procedures and expectations between the two domains caused frustration for patients as well as clinicians. One genetic counselor noted that the timeline for returning results was “certainly research-based” (NU). It was very slow, with many eMERGE-III sites experiencing delays of months to years in receiving results from the sequencing centers, compared with the more time-sensitive pace of clinical care, in part because the project was not primarily designed to benefit individual participants. The genetic counselor stated that this confused and aggravated many participants with whom she spoke. Participants did not understand the different timelines, expectations, or purposes of research and assumed that the relationship between them and their clinician and that between them and the investigator were the same.
A genetic counselor stressed that “we’re not following those three thousand patients [i.e., research participants] that we enrolled in eMERGE to see what’s going on with them, whereas a clinician is. There’s a different relationship” (NU). A clinician at a different site similarly said that it “feels crummy” (KP/UW) when the research nature of eMERGE-III prevented clinicians from following up with participants as they would with patients by providing clinically actionable results. “It’s a lot of work to actually get the information, but you have to remember, this is potentially life-saving information, so it’s really important,” noted one clinician (HU).
DISCUSSION
The reasons for the distinction between research and clinical activities are numerous, but some of these reasons are attributable to differences in goals, relationships, and activities. The primary goal of research is to create generalizable scientific knowledge, while the goal of clinical care is to benefit individual patients, a distinction that some scholars consider paramount.2,7 The relationship between investigator and research participant is dictated by different ethical and legal obligations and overseen by professional bodies separate from those that govern the relationship between clinician and patient. The latter relationship, characterized as fiduciary, is rooted in the trust patients place in their clinicians to hold their best interests as their own; such entrustment is at most only partial in the investigator–participant relationship and incurs fewer duties.15 The two activities are characterized by different standards, in particular the types and quantities of evidence marshalled in making decisions in their respective domains.1 Clinical care bases its practice on standards of care, professional and evidence-based guidelines, and malpractice adjudication, among other things,5 while research focuses on obtaining generalizable knowledge by using well-disciplined strategies.
The infrastructures for interactions with patients and research participants can diverge dramatically. The major goal of the health-care system is to deliver care to patients, and it is structured to that end. Clinical relationships are driven primarily by the needs of patient. Translational research, by contrast, must conform to protocols and collect data, which may or may not be needed for the care of the participants. This research is often conducted at least in part by investigators who have little or no interaction with participants.
The interviews analyzed above reveal several ethical issues that go to the heart of the research/clinical care dichotomy and of the translational research that seeks to bridge that divide. A primary goal of eMERGE-III was to learn how best to implement genomic science, focusing particularly on individuals who lack currently accepted clinical indications for testing. Some sites were particularly interested in assessing how the return of results could be achieved without relying entirely on genetics specialists, who represent a scarce human resource in health-care systems across the country.
Yet eMERGE-III was intrinsically limited in its ability to achieve these goals for a host of reasons. As we have reported elsewhere,16,17 the participants’ personal clinicians—the very ones who, once the tests were incorporated into routine care, presumably would order these tests themselves or would refer patients to other physicians for testing—often were not prepared or willing to deal with these results. Thus, eMERGE investigators in these studies often had to return results themselves, even though they typically did not have a personal relationship with participants as they would have with their own patients. It also meant dealing with the results for deceased participants, as they could be of value for surviving family members, and for pediatric participants who had reached the age of majority. The research infrastructure proved less well suited to disclosing results and providing counseling than the clinical enterprise, especially at the scale of this study, where results had to be returned to hundreds and even thousands of participants rather than at the one-on-one scale of disclosure that occurs in practice. These challenges in delivering results are particularly notable, since the eMERGE sites, as leaders in genomic medicine, are far better staffed and possess greater expertise than should be anticipated regarding most providers and health-care systems.
While the distinction between clinical care and research is well rehearsed in the literatures of ethics and law, many studies have shown that it does not appear to be so self-evident for lay patients1 or even some professionals.4 The division is also not so clear for clinicians who act as investigators, as we have seen in the eMERGE study. The mixing of these two roles can be challenging and confusing.5,8,18,19,20,21,22,23
The challenges of this research frequently triggered a clinical impulse in investigators, a type of moral distress that we suggest emerges from role conflict. The clinical impulse led investigators into a field of supererogatory actions in an effort more closely to approximate clinical care. For instance, many clinician-investigator teams in the eMERGE Consortium went to great lengths to change their protocols and to retrofit their consents to be allowed to return research results that they considered clinically actionable for their participants/patients. Similarly, some created delivery systems de novo to enable disclosure and counseling. In other words, they were driven to act as clinicians, rather than purely as researchers.
Limitations
The sites in the eMERGE Consortium, while all focused on assessing the impact of returning genomic results, varied in their research goals, the populations they recruited, and the infrastructure and processes they used for this research. In the current study, we sought to explore the ethical challenges that personnel experienced in conducting this research, issues to which our respondents may have been particularly attuned.
Conclusion
Translational research is important and, indeed, necessary for the progress of genomic medicine. From an ethical perspective, though, it is also inherently fraught. For it to be successful, translational research must mimic clinical practice, but for the many reasons iterated above, it does not and cannot do so perfectly. As has been addressed in the literature, philosophical and practical differences between research and clinical care make them noncommensurate in certain salient ways. Yet philosophical concerns are not the only ones at stake at the research/clinical interface. As this study has demonstrated, fundamental infrastructural problems in the rollout of large-scale genetic testing presented ethical challenges.
Returning results ethically requires informing participants in greater detail about the nature of the results they will receive, only some of which are well understood or clinically indicated. While achieving truly informed consent is challenging,24 one critical aspect of consent is relatively easy to address. It is now clear that many investigators intend to return results and that many patients expect to receive them. The revisions to the Common Rule require that this be specifically addressed in written informed consent.25 This must be done before the research is undertaken.
The systemic challenges of returning results that are not part of routine clinical screening, by contrast, are more difficult to tackle. A clinical staff with adequate training and infrastructure to respond to individual participants’ questions and concerns about results is essential. This will be a challenge, given how little many participants understand about genomic results17 and the scale of these projects. Another required element is humility in relying on such research studies to inform practice, since translational research inherently will not replicate the broad and variably resourced clinical domain in which these tests are to be used.
The heart of the problem is that translational research, by definition, asks whether a specific intervention should be deployed in a particular setting. Meanwhile, investigators who undertake such studies hypothesize—often based on practice and their experience in other settings—that at least some of their results will have clinical value. Their clinical impulse, then, can be expected, but the responses it elicits from investigators complicate the lessons to be learned from translational research.
References
Hall A, Alberg C, Hallowell N, Luheshi L Genomics and the boundary between research and clinical care and treatment. PHG Foundation. April 2014. http://www.phgfoundation.org/briefing_notes/303. Accessed 15 September 2019.
Beauchamp TL, Saghai Y. The historical foundations of the research-practice distinction in bioethics. Theor Med Bioeth. 2012;33:45–56.
Appelbaum PS, Roth LH, Lidz C. The therapeutic misconception: Informed consent in psychiatric research. Int J Law Psychiatry. 1982;5:319–329.
Lee SS-J, Kelley M, Cho MK, et al. Adrift in the gray zone: IRB perspectives on research in the learning health system. AJOB Empir Bioeth. 2016;7:125–134.
Wolf SM, Amendola LM, Berg JS, et al. Navigating the research–clinical interface in genomic medicine: analysis from the CSER Consortium. Genet Med. 2018;20:545–553.
Largent EA, Joffe S, Miller FG. Can research and care be ethically integrated? Hastings Cent Rep. 2011;41:37–46.
Kass NE, Faden RR, Goodman SN, Pronovost P, Tunis S, Beauchamp TL. The research‐treatment distinction: a problematic approach for determining which activities should have ethical oversight. Hastings Cent Rep. 2013;43:S4–S15.
Brody H, Miller FG. The research-clinical practice distinction, learning health systems, and relationships. Hastings Cent Rep. 2013;43:41–47.
Khoury MJ, Gwinn M, Yoon PW, Dowling N, Moore CA, Bradley L. The continuum of translation research in genomic medicine: how can we accelerate the appropriate integration of human genome discoveries into health care and disease prevention? Genet Med. 2007;9:665–674.
Wiesner GL, Rahm AK, Appelbaum PS, Aufox S, Bland S, Blout CL, et al. Returning results in the genomic era: initial experiences of the eMERGE network . J Pers Med. 2020;10:E30.
Wolf SM, Burke W, Koenig BA. Mapping the ethics of translational genomics: situating return of results and navigating the research-clinical divide. J Law Med Ethics. 2015;43:486–501.
Willis GB. Cognitive interviewing: a tool for improving questionnaire design. Thousand Oaks, CA: Sage; 2005.
Hsieh H-F, Shannon SE. Three approaches to qualitative content analysis. Qual Health Res. 2005;15:1277–1288.
Bradley EH, Curry LA, Devers KJ. Qualitative data analysis for health services research: developing taxonomy, themes, and theory. Health Serv Res. 2007;42:1758–1772.
Richardson HS, Belsky L. The ancillary-care responsibilities of medical researchers: an ethical framework for thinking about the clinical care that researchers owe their subjects. Hastings Cent Rep. 2004;34:25.
Pet DB, Holm IA, Williams JL, et al. Physicians’ perspectives on receiving unsolicited genomic results. Genet Med. 2019;21:311–318.
Halverson CM, Jones SH, Novak L, et al. What results should be returned from opportunistic screening in translational research? J Pers Med. 2020;10:13
Churchill LR. Physician–investigator/patient–subject: exploring the logic and the tension. J Med Philos. 1980;5:215–224.
Morin K, Rakatansky H, Riddick FA, et al. Managing conflicts of interest in the conduct of clinical trials. JAMA. 2002;287:78–84.
Joffe S, Miller FG. Bench to bedside: mapping the moral terrain of clinical research. Hastings Cent Rep. 2008;38:30–42.
Henderson GE, Churchill LR, Davis AM, et al. Clinical trials and medical care: defining the therapeutic misconception. PLoS Med. 2007;4:e324.
Jarvik GP, Amendola LM, Berg JS, et al. Return of genomic results to research participants: the floor, the ceiling, and the choices in between. Am J Hum Genet. 2014;94:818–826.
Burke W, Evans BJ, Jarvik GP. Return of results: ethical and legal distinctions between research and clinical care. Am J Med Genet C Semin Med Genet. 2014;166:105–111.
Flory JH, Wendler D. Emanuel EJ. Empirical issues in informed consent for research. In: Emmanuel EJ, Grady C, Crouch RA, Lie RK, Miller FG, Wendler D, editors. The Oxford textbook of clinical research ethics. Oxford: Oxford University Press; 2008. p. 645–660.
45 CFR § 46.116 - General requirements for informed consent. 1994.
Acknowledgements
This work was supported by two grants from the National Institute of Health: 5U01 HG008672-04 and 5R01 HG010004-02. This phase of the eMERGE Consortium was initiated and funded by the National Human Genome Research Institute (NHGRI) through the following grants: U01HG8657 (Group Health Cooperative/University of Washington); U01HG8685 (Brigham and Women’s Hospital); U01HG8672 (Vanderbilt University Medical Center); U01HG8666 (Cincinnati Children’s Hospital Medical Center); U01HG6379 (Mayo Clinic); U01HG8679 (Geisinger Clinic); U01HG8680 (Columbia University Health Sciences); U01HG8684 (Children’s Hospital of Philadelphia); U01HG8673 (Northwestern University); U01HG8701 (Vanderbilt University Medical Center serving as the Coordinating Center); U01HG8676 (Partners Healthcare/Broad Institute); and U01HG8664 (Baylor College of Medicine). The authors thank our interviewees for their time and assistance, including Carrie Blout, Adam Buchanan, John Connolly, Aileen Espinal, Alex Fedotov, Margaret Harr, Christie Hoell, David Kochan, Cindy Prows, James Ralston, Maureen Smith, Amy Sturm, Georgia Wiesner, and Erin Winkler, many of whom additionally provided very helpful, specific feedback on drafts of this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Halverson, C.M.E., Bland, S.T., Leppig, K.A. et al. Ethical conflicts in translational genetic research: lessons learned from the eMERGE-III experience. Genet Med 22, 1667–1672 (2020). https://doi.org/10.1038/s41436-020-0863-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0863-9
Keywords
This article is cited by
-
A practical checklist for return of results from genomic research in the European context
European Journal of Human Genetics (2023)
-
My Research Results: a program to facilitate return of clinically actionable genomic research findings
European Journal of Human Genetics (2022)
