
Abstract
Purpose
This study investigated the diagnostic utility of nontargeted genomic testing in patients with pediatric heart disease.
Methods
We analyzed genome sequencing data of 111 families with cardiac lesions for rare, disease-associated variation.
Results
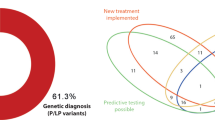
In 14 families (12.6%), we identified causative variants: seven were de novo (ANKRD11, KMT2D, NR2F2, POGZ, PTPN11, PURA, SALL1) and six were inherited from parents with no or subclinical heart phenotypes (FLT4, DNAH9, MYH11, NEXMIF, NIPBL, PTPN11). Outcome of the testing was associated with the presence of extracardiac features (p = 0.02), but not a positive family history for cardiac lesions (p = 0.67). We also report novel plausible gene–disease associations for tetralogy of Fallot/pulmonary stenosis (CDC42BPA, FGD5), hypoplastic left or right heart (SMARCC1, TLN2, TRPM4, VASP), congenitally corrected transposition of the great arteries (UBXN10), and early-onset cardiomyopathy (TPCN1). The identified candidate genes have critical functions in heart development, such as angiogenesis, mechanotransduction, regulation of heart size, chromatin remodeling, or ciliogenesis.
Conclusion
This data set demonstrates the diagnostic and scientific value of genome sequencing in pediatric heart disease, anticipating its role as a first-tier diagnostic test. The genetic heterogeneity will necessitate large-scale genomic initiatives for delineating novel gene–disease associations.
Similar content being viewed by others

INTRODUCTION
Congenital heart disease (CHD) describes a heterogeneous set of disorders that affect the structure or function of the developing heart. With a birth prevalence of 1–3%, CHD is the most common congenital anomaly in humans.1 Childhood cardiomyopathies are progressive disorders and a common cause of heart failure in children.2 The causes of pediatric heart disease are diverse and often multifactorial. Evidence for major genetic contributions come from familial recurrence rates, twin studies, and a higher incidence in consanguineous populations.3,4 Mendelian forms of idiopathic CHD are considered rare, and many of the known loci are associated with incompletely penetrant, variable cardiac, and extracardiac manifestations. Clinical genetic assessments are not systematically offered to families with cardiac lesions, and there are no formal diagnostic testing protocols. Limited accession of genetic services and hypothesis-driven approaches may result in etiological underdiagnoses and/or diagnostic odysseys.
High-throughput (exome or genome) sequencing studies of cohorts with CHD had reported remarkably disparate diagnostic rates (5.2–43.3%), which also correlated with the stringency in variant interpretation.5,6,7,8 Compared with less comprehensive techniques, genome sequencing allows for an unbiased analysis of most types of genomic variation, and had a higher yield than standard of care genetic testing in clinically heterogeneous cohorts.9 However, variant interpretation may be challenging, particularly for sporadic disease with limited genotype–phenotype correlations and incomplete penetrance.
The Cardiac Genome Clinic was established to investigate the utility of genome sequencing in families with pediatric heart disease. As part of a pilot study, we obtained genome sequencing data of 111 unrelated probands (n = 107 sequenced as parent–child trios/quartets/extended families), and systematically analyzed for rare, predicted damaging variation (single-nucleotide variants, insertions/deletions and structural variants). Variants in disease-associated genes were interpreted according to standard guidelines,10 and novel candidate genes were prioritized according to their biological plausibility.
MATERIALS AND METHODS
Study participants
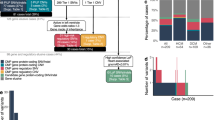
The study was approved by the Research Ethics Board at The Hospital for Sick Children (REB #1000053844). Informed consent was obtained from all probands and family members. Study participants originated from a cohort of families with pediatric heart disease, recruited through the Ted Rogers Cardiac Genome Clinic at a single site, The Hospital for Sick Children, Division of Cardiology (from January 2017 to December 2018; Fig. 1, S1). By study design, families with laterality defects, outflow tract obstructions, or cardiomyopathies were preferentially enrolled. Exclusion criteria were known syndromes, metabolic diseases, or medical conditions leading to secondary heart failure. Phenotype data were entered into PhenoTips (https://phenotips.org/), using the Human Phenotype Ontology (https://hpo.jax.org/). If possible, we sequenced genomes of parent–child trios/quartets (n = 103), or multiple affected relatives (n = 4), resulting in a total of 328 sequenced individuals from 111 families.

Families with pediatric heart disease (n = 111) were recruited through the Division of Cardiology at The Hospital for Sick Children. The genome sequencing data was analyzed for small nucleotide and structural variation. Clinically relevant variants were returned to participants per consent. CNV copy-number variant.
Genome sequencing and annotation
DNA was sequenced on the Illumina HiSeq X system at The Centre for Applied Genomics (TCAG) in Toronto, Canada (details on sequencing and data analysis as supplementary information). Genome sequencing was performed under a research protocol, not as a validated clinical test. Population allele frequencies were derived from 1000 Genomes (https://www.internationalgenome.org/), ExAC (http://exac.broadinstitute.org/), and gnomAD (https://gnomad.broadinstitute.org/). Gene constraint metrics were derived from ExAC (probability of loss-of-function intolerance; pLI) and gnomAD (pLI, observed over expected loss-of-function variants; o/e). Variant information was queried from PubMed (https://www.ncbi.nlm.nih.gov/pubmed/), DECIPHER (https://decipher.sanger.ac.uk/), the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php), and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
Variant prioritization and interpretation
We analyzed the data for various types of genomic variation (small variants affecting single genes, copy number, and other structural aberrations) and Mendelian inheritance patterns (de novo, recessive, and dominantly inherited, also considering incomplete penetrance; Fig. 2). Inherited variants were prioritized according to (1) cosegregation with disease, (2) previous reports in cardiovascular disease, (3) predicted loss-of-function of constrained genes (ExAC pLI ≥0.9), and (4) predicted damaging effects in CHD genes (http://chdgene.victorchang.edu.au/).8 Variants in genes known to be associated with cardiac disease were interpreted in accordance with clinical standards and guidelines of the American College of Medical Genetics and Genomics (ACMG).10 Likely pathogenic, pathogenic, and uncertain variants were reviewed by a clinical geneticist, a genetic counselor, and a cardiologist in the context of the phenotype and family history. For the diagnostic yield, we considered variants deemed “causative” for the CHD by the clinical assessment. For novel candidate genes, we assessed the biological and experimental plausibility based on a literature review. Copy-number variants (CNVs) were interpreted regarding a known or potential role in cardiovascular disorders. Variants of interest were confirmed through Sanger sequencing or clinical microarrays. Relevant findings were reported back to the families through a clinical geneticist and a genetic counselor, and were sent for clinical validation.

Different inheritance patterns were considered to allow a comprehensive assessment of genomic variation. CHD congenital heart disease, HGMD Human Gene Mutation Database, LOF loss of function.
RESULTS
Cohort characteristics
We prospectively recruited 111 families with congenital heart disease or childhood-onset cardiomyopathies. Of those, 53 probands (47.7%) had extracardiac features, defined as other major malformations, intellectual disability, autism, global developmental delay, or growth deficits not attributable to heart failure. Thirteen families (11.7%) reported relatives with clinically relevant cardiac lesions, two probands (1.8%) had a parent with bicuspid aortic valve, and four parents (3.6%) were consanguineous (Table 1, S1). Ninety families (81.1%) were formally assessed by a clinical geneticist, 85 had parental echocardiography (n = 78 biparental; 70.3%), and 71 (64.0%) had negative standard of care genetic testing (such as chromosomal microarrays, targeted gene/panel testing, clinical exome sequencing; Table S1). The spectrum of primary cardiac phenotypes is displayed in Table 1, phenotypic details in Table S1.
Clinically relevant variants
To assess the diagnostic utility of genome sequencing in children with cardiac disease, we interpreted the data for clinically relevant variants. We identified causative variants in 14 of 111 families (12.6%); Table 2. Ten of the affected genes were found on a curated list of 107 high-confidence CHD-associated genes (http://chdgene.victorchang.edu.au/; December 2019). Eleven diagnoses were made in patients with extracardiac features (11/53 vs. 3/58; Fisher’s exact test (FET): p = 0.02), and two in patients with familial heart defects (2/13 vs. 12/98; FET: p = 0.67).
Small nucleotide variants
Seven individuals with prominent extracardiac anomalies and developmental delay had disease-causing de novo variants, such as p.(Pro1747Argfs*49) in ANKRD11 (KBG syndrome), or p.(Arg5225Cys) in KMT2D (Kabuki syndrome). Both genes are associated with highly penetrant congenital heart defects. In a patient with ventricular septal defect (VSD), aortic coarctation, and neurological symptoms, we identified a de novo missense substitution p.(Val224Asp) in the ligand binding domain of NR2F2. Missense variants in NR2F2 were associated with CHD (particularly septal defects) and a broad spectrum of associated anomalies.11 A de novo variant p.(Glu1135Argfs*3) in POGZ was found in a proband with hypoplastic left heart syndrome (HLHS) and developmental delay, supporting the gene’s pleiotropic effects in brain and heart development.12,13 A proband with VSD, developmental delay, hypotonia, respiratory issues, and growth anomalies had two de novo, recurrent variants p.(Phe271del) in PURA and p.(Gly132Asp) in PTEN. Both defects contribute to the phenotype, and a minority of patients with PURA-related disorders present with structural heart defects.14
In other cases of apparently sporadic CHD, pathogenic nucleotide variants were inherited from parents with no or subclinical heart disease (n = 5). A frameshift deletion p.(Pro30Argfs*3) in FLT4 was identified in a patient with tetralogy of Fallot (TOF) and her unaffected mother.15 FLT4 haploinsufficiency was recently associated with incompletely penetrant nonsyndromic TOF.15,16,17 A patient with aortic stenosis, valve dysplasia, and developmental delay had a variant p.(Gly501Valfs*4) in NEXMIF, which was X-linked inherited from the mother with mild intellectual disability and epilepsy. Cardiac defects are not common for NEXMIF-related disease, but valve dysfunctions (pulmonary stenosis, mitral insufficiency) were infrequently reported.18 In a patient with hypoplastic right heart, unbalanced septal defect, developmental delay, and borderline microcephaly, we identified a pathogenic NIPBL variant c.771+1G>A for Cornelia de Lange syndrome. The variant was inherited from the mother, who had short stature, small hands, and delayed menarche, but normal cognitive and cardiac presentation, indicating variable expressivity. The broad spectrum of cardiac lesions associated with NIPBL haploinsufficiency likely results from subtle transcriptional dysregulations of hundreds of genes.19 A patient with dysplastic aortic and pulmonary valve and borderline short stature was identified with a maternally inherited PTPN11 variant p.(Lys70Arg) for Noonan syndrome. The mother was considered healthy; however, a research echo at the time of study enrollment showed decreased ventricular function of unknown origin. A paternally inherited, pathogenic MYH11 variant c.4578+1G>A, resulting in an in-frame loss of 71 amino acids,20 was identified in a proband with patent ductus arteriosus (PDA). Cosegregation testing in four paternal relatives with PDA and a grandfather with aortic disease in his eighties could not be performed. The same protein change was reported in an unrelated family with familial PDA.21 Further phenotype–genotype correlations are required to provide risk estimates for aortic disease in such families. In a female patient with dextrocardia and unbalanced atrioventricular septal defect, we identified compound heterozygous missense variants in DNAH9, a gene recently associated with laterality defects.22 Both variants p.(Asp1474Gly) and (Ile4415Thr) were rare and predicted to be damaging. Though pathogenicity could not be established from a molecular perspective, the variants were considered causative due to mild respiratory issues in the proband, and ultrastructural ciliary abnormalities on electron microscopy of nasal mucosa performed after clinical reassessment.
Structural variants
Genome sequencing analyses identified two pathogenic CNVs: A 138-kb deletion, including NOTCH1, was identified in a proband with TOF and pulmonary atresia, but no obvious other features of Adams–Oliver syndrome. Cosegregation studies in three family members with VSD or TOF could not be performed. A patient with interrupted aortic arch, large VSD, short stature, and dysplastic ears was found to have a de novo 4.1-Mb deletion including SALL1, causing Townes–Brocks syndrome.
Variants of uncertain relevance
Variants that did not meet criteria for pathogenicity were identified in additional families. A family with hypertrophic cardiomyopathy had a predicted damaging, cosegregating missense substitution p.(Gly2080Arg) in FLNC, located in a previously disease-associated transmembrane domain.23 A patient with dextrocardia and a complex heart defect, short stature, and failure to thrive was identified with rare, compound heterozygous missense variants p.(Thr331Ala) and (Phe3591Leu) in DNAH8, a gene associated with primary ciliary dyskinesia. This proband also had a maternally inherited 8.3-Mb deletion at 3p11.2-3p12.3 (including ROBO1, a candidate gene for TOF and septal defects24). A paternally inherited 594-kb microdeletion at 2p13.1-2p12, encompassing 21 coding genes, was found in a patient with aortic coarctation and bicuspid aortic valve, and a patient with hypoplastic right heart, tricuspid valve dysplasia, septal defect, and mild intellectual disability had a maternally inherited structural aberration involving a 4.3-Mb deletion at 3p26.1-3pter, and a 1.8-Mb duplication at 3p26.1. A 2.4-Mb duplication at 15q13.2-15q13.3 in a proband with interrupted aortic arch, aortic stenosis, and VSD was inherited from the healthy father; however, six largely overlapping duplications of 2–2.5 Mb in the DECIPHER database had occurred de novo, indicating a potential disease locus.
For two variants, though they were classified as likely pathogenic, the causal link to the presenting cardiac condition remained uncertain: a de novo 9-kb deletion of the first exon and promoter region of DSG2 was identified in a 6-year-old proband with atrial septal defect and dilated right ventricle, but was considered a secondary finding. A pathogenic PTEN variant p.(Arg15Ser) segregated in a proband with aortic coarctation and his father with bicuspid aortic valve, yet was not deemed causative for the heart defect according to present knowledge. When applying less stringent variant interpretation principles, the yield of potentially relevant variants could become higher (up to 19.8%; 22/111; Table 2).
Novel candidate genes
We also analyzed the data for biologically plausible novel gene–disease associations (Table 3; supplementary information). In this respect, we and others15,16,17 had recently reported an association of vascular endothelial growth factor (VEGF) signaling gene(s) and TOF, including two novel variants identified in this cohort: a missense change p.(Ala1030Thr) in the protein kinase domain of KDR, and a stopgain variant p.(Arg766*) in IQGAP1.15 The disease relevance of IQGAP1 was affirmed by an exome sequencing study, which reported two de novo loss-of-function variants in fetuses with TOF or transposition of the great arteries, respectively.7 Consistent with our recently published data, we identified an FGD5 stopgain variant p.(Glu322*) in a proband with critical pulmonary stenosis and dysplastic valve, adding evidence for an involvement of the VEGF signaling pathway in pulmonary valve development. We also identified a de novo stopgain variant p.(Gln24*) in CDC42BPA in a proband with sporadic TOF and right aortic arch. The encoded protein has roles in cytoskeletal remodeling and cell migration, and is a binding partner of CDC42, a GTPase essential for VEGF signaling and developmental processes.25,26
In two unrelated families with HLHS, we identified compound heterozygous variants in VASP or TLN2, respectively (Table 3). Both genes are involved in mechanotransduction of developing cardiomyocytes, linking mechanical strain and cardiac remodeling. Two siblings with hypoplastic right heart had an apparently de novo missense variant p.(Arg171Gln) in TRPM4, though one sequencing read suggested potential low-level paternal mosaicism (supplementary information). Trpm4 is involved in the determination of murine heart size, potentially through a regulation of myocyte proliferation during fetal development.27 In a patient with atrioventricular septal defect, mild left ventricular hypoplasia, and extracardiac features, we identified a frameshift insertion p.(Lys615Ilefs*49) in SMARCC1, a highly constrained gene encoding a core subunit of the SWI/SNF chromatin remodeling complex. The variant was inherited from the father, diagnosed in adulthood with bicuspid aortic valve. Smarcc1 knockdown in zebrafish was associated with variable multiorgan defects, whereby cardiovascular anomalies were the most penetrant feature.28 In humans, haploinsufficiency for other SWI/SNF subunits is associated with developmental disorders, including heart defects: ARID1A, ARID1B, ARID2, SMARCA4, SMARCB1, SMARCC2, SMARCE1, ACTL6A, and DPF2.29
A TPCN1 missense variant p.(Arg199Gln) had occurred de novo in a patient with early-onset, devastating dilated cardiomyopathy. TPCN1 encodes a lysosomal ion channel (i.e., Ca2+), and increased expression was associated with dilated cardiomyopathy and heart failure.30 A homozygous frameshift deletion p.(Ser159Argfs*44) in UBXN10 was found in a patient with congenitally corrected transposition of the great arteries, VSD, and pulmonary stenosis. The encoded protein is required for ciliogenesis, and Ubxn10 depletion caused cardiac laterality defects in zebrafish.31 Predicted loss-of-function variants in GMDS (Ebstein anomaly), SRPK2 (HLHS), and TOP2A (HLHS) were also considered candidates for the cardiac phenotypes (supplementary information).
DISCUSSION
The value of genetic testing in severe, congenital disorders is widely recognized, as it may specify recurrence risks and potential comorbidities, and will ultimately support optimized clinical management and outcomes.32 Implementation of standardized genetic testing protocols in infants with critical CHD had resulted in higher diagnostic rates and cost efficiency.33 Nonetheless, there is presently no consensus on appropriate genetic testing in families with cardiac lesions, and the genomic architecture is fairly unknown.
This study set out to investigate the diagnostic utility of genome sequencing in a cohort of pediatric heart disease. Although many of the families had undergone prior genetic testing (Table S1), genome sequencing identified a disease-causing variant in 14 of 111 probands (12.6%). The majority of genes were associated with “syndromic disease” (e.g., ANKRD11, KMT2D, and POGZ) and were detected in individuals with extracardiac features. Particularly in young children, associated features may be nonspecific, or erroneously attributed to the cardiac lesion. A further aspect impeding the recognition of genetic syndromes is the tremendous clinical variation even of well-defined disorders. This was evident as pathogenic alleles were inherited from ostensibly healthy parents (e.g., MYH11, NIPBL, and PTPN11). By contrast, six of seven variants primarily associated with transcriptional regulation/chromatin organization were de novo, potentially due to pleiotropy and a higher rate of extracardiac features12 (Fig. S2). Immediate implications on patient management and genetic counseling were related to variants in ANKRD11, DNAH9, DSG2, KMT2D, MYH11, NEXMIF, NIPBL, NOTCH1, PURA, POGZ, PTEN, PTPN11 (×2), and SALL1.
The variety of clinically unexpected—or formerly undetected—findings supports a role of genome sequencing as a first-tier diagnostic test in patients with CHD.34 Genome sequencing was previously shown to have adequate coverage for clinically relevant gene sets,9 and can overcome several technical limitations of exome sequencing and chromosomal microarray analysis, particularly for small structural variations.35 In this study, we demonstrate the detection and fine-mapping of a wide size range of potentially disease-related CNVs (9.1 kb to 8.3 Mb), with reliable detection rates previously shown to exceed microarrays.36
For the majority of individuals in this study, the etiology of their heart defects remained unknown. This held true even for some families where a genetic condition was strongly suspected. Though genome sequencing has the potential to capture most types of interindividual genomic differences, our abilities to identify those that are disease-relevant lag behind. This particularly applies for noncoding regions and synonymous variants, but also for the multitude of nonsynonymous alterations with uncertain effects upon protein function. Genomic curation largely depends on manual data review, even when applying assisting software to partially automate the process.37 Variant interpretation in congenital heart disease can be particularly challenging due to limited genotype–phenotype correlations and incomplete penetrance. Disease associations and functional studies from the literature need to be critically reviewed and potentially reassessed. Even when applying established guidelines,10 the evaluation of genomic variation is subjective and potentially discordant among analysts.38 With stringent application of the ACMG guidelines, we consider our interpretation to be conservative, compared with CHD studies with higher diagnostic yields5,8 (Table S3). As for other heterogeneous diseases with incomplete penetrance, the contribution of rare, inherited variants is most likely underestimated. A comprehensive delineation of the genomic spectrum will involve statistical approaches and functional assays. The full potential of genomic data analysis will evolve prospectively, and the yield is expected to increase accordingly.
By study design, findings from this cohort were not transferable to a general CHD population. We enriched the cohort for more complex cardiac conditions with a supposedly stronger genetic etiology, such as outflow tract anomalies and single functional ventricles,4 though we also identified relevant diagnoses in families with isolated lesions. On the other hand, many probands had negative clinical genetic testing prior to study enrollment (Table S1), which may account for the relatively low numbers of pathogenic CNVs, for instance.39 In this study, the outcome of the testing was associated with the presence of extracardiac features, but not a positive family history for clinically relevant CHD. As other data suggested the diagnostic yield to also depend on the fraction of familial cases,5 larger studies will need to refine which patients will most likely benefit from genomic testing.
In the attempt to disentangle the genetic basis of pediatric heart disease, our systematic analysis revealed possible new candidate genes (Table 3). We prioritized variants based on known disease mechanisms in cardiac development, such as disruptions of critical biological functions and pathways, or altered dosage of constrained signaling genes.3 However, the validation of novel gene–disease associations is challenged by the genetic heterogeneity, as recurrence in unrelated families, or ideally significant enrichment on a variant or gene level, would require very large cohorts. Small pedigrees and incomplete penetrance (e.g., through multilocus inheritance with rare and common modifiers40) further impede classical linkage or cosegregation evidence. The functional assessment of candidate genes in animal or cellular models is time-consuming, and transferability to human heart development is limited. Sharing potential (unverified) gene–disease associations, e.g., in databases and the scientific literature, is therefore evidentially valuable for the assembly of independent evidence and the design of follow-up studies.
Our data outline the diagnostic and scientific utility of comprehensive (nontargeted) genetic testing in families with pediatric heart disease, and anticipate that genome sequencing will ultimately become a first-tier diagnostic test. Many cardiac disease–gene associations are likely yet to be unraveled, and this attempt will require large-scale genomic initiatives and interdisciplinary efforts for experimental validations.
References
Pierpont ME, Brueckner M, Chung WK, et al. Genetic basis for congenital heart disease: revisited: a scientific statement from the American Heart Association. Circulation. 2018;138:e653–e711.
Vasilescu C, Ojala TH, Brilhante V, et al. Genetic basis of severe childhood-onset cardiomyopathies. J Am Coll Cardiol. 2018;72:2324–2338.
Fahed AC, Gelb BD, Seidman JG, Seidman CE. Genetics of congenital heart disease: the glass half empty. Circ Res. 2013;112:707–720.
Oyen N, Poulsen G, Boyd HA, Wohlfahrt J, Jensen PK, Melbye M. Recurrence of congenital heart defects in families. Circulation. 2009;120:295–301.
Alankarage D, Ip E, Szot JO, et al. Identification of clinically actionable variants from genome sequencing of families with congenital heart disease. Genet Med. 2019;21:1111–1120.
Hauser NS, Solomon BD, Vilboux T, Khromykh A, Baveja R, Bodian DL. Experience with genomic sequencing in pediatric patients with congenital cardiac defects in a large community hospital. Mol Genet Genomic Med. 2018;6:200–212.
Petrovski S, Aggarwal V, Giordano JL, et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet. 2019;393:758–767.
Szot JO, Cuny H, Blue GM, et al. A screening approach to identify clinically actionable variants causing congenital heart disease in exome data. Circ Genom Precis Med. 2018;11:e001978.
Lionel AC, Costain G, Monfared N, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med. 2018;20:435–443.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Al Turki S, Manickaraj AK, Mercer CL, et al. Rare variants in NR2F2 cause congenital heart defects in humans. Am J Hum Genet. 2014;94:574–585.
Homsy J, Zaidi S, Shen Y, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350:1262–1266.
Stessman HAF, Willemsen MH, Fenckova M, et al. Disruption of POGZ is associated with intellectual disability and autism spectrum disorders. Am J Hum Genet. 2016;98:541–552.
Reijnders MRF, Leventer RJ. Lee BH, et al.PURA-related neurodevelopmental disorders. In: Adam MP, Ardinger HH, Pagon R et al., editors. GeneReviews. Seattle, WA: University of Washington; 1993.
Reuter MS, Jobling R, Chaturvedi RR, et al. Haploinsufficiency of vascular endothelial growth factor related signaling genes is associated with tetralogy of Fallot. Genet Med. 2019;21:1001–1007.
Jin SC, Homsy J, Zaidi S, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49:1593–1601.
Page DJ, Miossec MJ, Williams SG, et al. Whole exome sequencing reveals the major genetic contributors to nonsyndromic tetralogy of Fallot. Circ Res. 2019;124:553–563.
Lorenzo M, Stolte-Dijkstra I, van Rheenen P, Smith RG, Scheers T, Walia JS. Clinical spectrum of KIAA2022 pathogenic variants in males: case report of two boys with KIAA2022 pathogenic variants and review of the literature. Am J Med Genet A. 2018;176:1455–1462.
Mills JA, Herrera PS, Kaur M, et al. NIPBL(+/-) haploinsufficiency reveals a constellation of transcriptome disruptions in the pluripotent and cardiac states. Sci Rep. 2018;8:1056.
Zhu L, Vranckx R, Khau Van Kien P, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38:343–349.
LaHaye S, Corsmeier D, Basu M, et al. Utilization of whole exome sequencing to identify causative mutations in familial congenital heart disease. Circ Cardiovasc Genet. 2016;9:320–329.
Loges NT, Antony D, Maver A, et al. Recessive DNAH9 loss-of-function mutations cause laterality defects and subtle respiratory ciliary-beating defects. Am J Hum Genet. 2018;103:995–1008.
Roldan-Sevilla A, Palomino-Doza J, de Juan J, et al. Missense mutations in the FLNC gene causing familial restrictive cardiomyopathy. Circ Genom Precis Med. 2019;12:e002388.
Kruszka P, Tanpaiboon P, Neas K, et al. Loss of function in ROBO1 is associated with tetralogy of Fallot and septal defects. J Med Genet. 2017;54:825–829.
Martinelli S, Krumbach OHF, Pantaleoni F, et al. Functional dysregulation of CDC42 causes diverse developmental phenotypes. Am J Hum Genet. 2018;102:309–320.
Basagiannis D, Zografou S, Murphy C, et al. VEGF induces signalling and angiogenesis by directing VEGFR2 internalisation through macropinocytosis. J Cell Sci. 2016;129:4091–4104.
Demion M, Thireau J, Gueffier M, et al. Trpm4 gene invalidation leads to cardiac hypertrophy and electrophysiological alterations. PLoS One. 2014;9:e115256.
Scott CA, Marsden AN, Rebagliati MR, et al. Nuclear/cytoplasmic transport defects in BBS6 underlie congenital heart disease through perturbation of a chromatin remodeling protein. PLoS Genet. 2017;13:e1006936.
Machol K, Rousseau J, Ehresmann S, et al. Expanding the spectrum of BAF-related disorders: de novo variants in SMARCC2 cause a syndrome with intellectual disability and developmental delay. Am J Hum Genet. 2019;104:164–178.
Garcia-Rua V, Otero MF, Lear PV, et al. Increased expression of fatty-acid and calcium metabolism genes in failing human heart. PLoS ONE. 2012;7:e37505.
Raman M, Sergeev M, Garnaas M, et al. Systematic proteomics of the VCP-UBXD adaptor network identifies a role for UBXN10 in regulating ciliogenesis. Nat Cell Biol. 2015;17:1356–1369.
Wright CF, FitzPatrick DR, Firth HV. Paediatric genomics: diagnosing rare disease in children. Nat Rev Genet. 2018;19:253–268.
Geddes GC, Basel D, Frommelt P, Kinney A, Earing M. Genetic testing protocol reduces costs and increases rate of genetic diagnosis in infants with congenital heart disease. Pediatr Cardiol. 2017;38:1465–1470.
De Backer J, Bondue A, Budts W. et al. Genetic counselling and testing in adults with congenital heart disease: a consensus document of the ESC Working Group of Grown-Up Congenital Heart Disease, the ESC Working Group on Aorta and Peripheral Vascular Disease and the European Society of Human Genetics. Eur J Prev Cardiol. 2019 Jun 11; https://doi.org/10.1177/2047487319854552 [Epub ahead of print].
Bick D, Jones M, Taylor SL, Taft RJ, Belmont J. Case for genome sequencing in infants and children with rare, undiagnosed or genetic diseases. J Med Genet. 2019;56:783–791.
Trost B, Walker S, Wang Z, et al. A comprehensive workflow for read depth-based identification of copy-number variation from whole-genome sequence data. Am J Hum Genet. 2018;102:142–155.
Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet. 2017;100:267–280.
Amendola LM, Jarvik GP, Leo MC, et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet. 2016;98:1067–1076.
Costain G, Silversides CK, Bassett AS. The importance of copy number variation in congenital heart disease. NPJ Genom Med. 2016;1:16031.
Gifford CA, Ranade SS, Samarakoon R, et al. Oligogenic inheritance of a human heart disease involving a genetic modifier. Science. 2019;364:865–870.
Acknowledgements
We thank the patients and their families for participating in this study. We thank the bioinformatics staff of The Centre for Applied Genomics (a node of CGEn) for assistance with data analysis. This study made use of data generated by the DECIPHER community, and the Genome Aggregation Database (gnomAD). This work was funded by the Ted Rogers Centre for Heart Research. S.W.S. is funded by the GlaxoSmithKline-CIHR Chair in Genome Sciences at the University of Toronto and The Hospital for Sick Children.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Reuter, M.S., Chaturvedi, R.R., Liston, E. et al. The Cardiac Genome Clinic: implementing genome sequencing in pediatric heart disease. Genet Med 22, 1015–1024 (2020). https://doi.org/10.1038/s41436-020-0757-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0757-x
Keywords
This article is cited by
-
Insights into the ANKRD11 variants and short-stature phenotype through literature review and ClinVar database search
Orphanet Journal of Rare Diseases (2024)
-
Association study of FLT4 and HYDIN single nucleotide polymorphisms with atrial septal defect susceptibility in the Han Chinese population of Southwest China
Italian Journal of Pediatrics (2024)
-
The chromatin regulator Ankrd11 controls cardiac neural crest cell-mediated outflow tract remodeling and heart function
Nature Communications (2024)
-
Expanding the phenotypic spectrum of NOTCH1 variants: clinical manifestations in families with congenital heart disease
European Journal of Human Genetics (2024)
-
White-Sutton syndrome and congenital heart disease: case report and literature review
BMC Pediatrics (2023)
