
Abstract
Purpose
Germline pathogenic variants are estimated to affect 3–5% of renal cell carcinoma (RCC) patients. However, higher mutational prevalence in non–clear cell RCC (non-ccRCC) and advanced disease has been suggested.
Methods
To clarify the prevalence of pathogenic germline variants in metastatic RCC, we sequenced 29 cancer susceptibility genes in 294 unselected metastatic RCC cases plus 21 patients with clinical hereditary features. In 145 tumors, genes frequently mutated in RCC were sequenced and methylation was assessed in selected cases.
Results
Germline variants in RCC predisposition genes (FH, VHL) were detected in 1.4% of the unselected metastatic patients, with higher frequency in non-ccRCC versus ccRCC (6.4% and 0.4%; P = 0.0025) and in younger patients (P = 0.036). Among the 315 studied patients, 14% of non–type 1 papillary cases (4 of 28), all metastatic <1 year after diagnosis, carried a FH germline variant with loss of heterozygosity and tumor genome hypermethylation. Variants in other cancer-associated genes (e.g., MUTYH, BRCA2, CHEK2) occurred in 5.1% of the unselected series, with unclear significance for RCC.
Conclusion
Our findings confirm a high prevalence of pathogenic germline variants in RCC predisposition genes in metastatic non-ccRCC, and highlight that metastatic patients with papillary type 2 or unconventional histologies compatible with FH would benefit from genetic screening.
Similar content being viewed by others

INTRODUCTION
Kidney cancer is the third most common urologic cancer and accounts for 2–3% of adult malignancies.1 Renal cell carcinoma (RCC) is a heterogeneous disease with different histological subtypes, in which clear cell (ccRCC), papillary (pRCC) types 1 and 2, and chromophobe (chRCC) represent more than 90% of all cases. Other subtypes consist of rare histological variants, such as collecting duct or medullary RCC, together with tumors that remain unclassified.2
While kidney tumors are mainly sporadic, the hereditary forms are estimated to account for 3–5% of all RCC cases.3 Familial cases are associated with pathogenic germline variants in BAP1, FLCN, FH, MET, PTEN, SDHB, TSC1, TSC2, or VHL, which increase the risk of different RCC histologic subtypes.4 For example, VHL variants are associated with ccRCC, MET with pRCC type 1, FLCN with mixed oncocytoma and/or chRCC, FH with pRCC type 2 but also collecting tube tumors or those with mixed architectural pattern.5,6
Hereditary forms tend to occur at an earlier age and can present multifocal or bilateral tumors.7 Large population-based studies found a significantly higher RCC risk for the family of affected individuals and suggested germline variants to be significantly involved in apparently sporadic RCC.8 A large retrospective study with 1,235 RCC patients selected for genetic screening found that 6% harbored a pathogenic variant in a high-risk RCC gene6 and the same percentage was found by Wu et al. in patients with early RCC onset.9 Regarding advanced disease, a recent study found that 12% of non-ccRCC patients carried germline variants in a RCC predisposition gene and that CHEK2 variants were present in 5% of ccRCC cases, suggesting that germline variants may be more prevalent in advanced RCC than in early-stage disease,10 as it occurs in other cancer types.11,12 However, these results remain to be further investigated in different patient populations with particular molecular and clinical characteristics.
Therefore, the prevalence of pathogenic germline variants in RCC varies among published cohorts, and the impact of histologic subtypes or advanced disease has not been fully elucidated. In this study, we performed a germline and somatic genetic characterization of a large number of Spanish metastatic RCC patients, to determine the prevalence and spectrum of inherited variants in metastatic disease.
MATERIALS AND METHODS
Patient characteristics
Cohort of unselected metastatic patients
RCC patients were recruited consecutively through an observational multicenter study from the Spanish Oncology Genitourinary Group (SOGUG). From these, peripheral blood or saliva samples were available from the 294 cases included in this study. Of these patients, 282 had been described in studies with other purposes,13,14 but none of them were subjected to previous germline mutational screening. The only inclusion criteria was that patients were diagnosed with RCC, were at least 18 years of age, and attended a medical oncology unit with metastatic disease to receive systemic treatment. This study is based on Spanish Caucasians as all patients were recruited in the oncology departments of hospitals in Spain. Among 104 patients for whom ethnicity data were collected, 97% were Spanish Caucasians. Formalin-fixed paraffin embedded (FFPE) tumor material was available from 145 of the patients.
Cohort with clinical RCC hereditary features
Twenty-one patients diagnosed with RCC and at least one of the following criteria: RCC diagnosis at 46 years or younger, multifocal/bilateral disease, or family history of RCC, were recruited at the Familial and Hereditary Cancer Unit of the Hospital 12 de Octubre. Peripheral blood and saliva samples were collected from each patient.
Genetic screening and variant interpretation
Genomic DNA was purified from whole blood or saliva samples by using Maxwell® RSC Blood DNA Kit (Promega) or prepIT·L2P purifier reagent (DNA Genotek), respectively. Primary tumor FFPE DNA was isolated from samples with at least 70% tumor content using Maxwell® RSC DNA FFPE Kit (Promega). DNA concentration was quantified using Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific).
Germline DNA was sequenced with a gene panel targeting the coding region of 29 cancer susceptibility genes including RCC syndromic genes (BAP1, FLCN, FH, MET, MITF, PTEN, SDHA, SDHB, SDHC, SDHD, TSC1, TSC2, and VHL) and other cancer-related genes (hereinafter referred to as “hereditary cancer panel”; Supplementary Table 1). Tumor samples were sequenced using a panel targeting the coding region of 43 genes found frequently mutated in sporadic RCC (hereinafter “somatic cancer panel”; Supplementary Table 1). In cases with a pathogenic germline variant and tumor available, tumor DNA was also sequenced with the hereditary cancer panel for detection of second hits and loss of heterozygosity (LOH).
For library preparation SeqCap EZ Choice Enrichment Kit (Roche) was used according to manufacturer’s instructions. In brief, 250–500 ng of DNA were used for the capture-based target enrichment. Sequencing was performed in a HiSeq sequencer (Illumina) configured to generate 100 bp paired-end reads. A total of 315 germline unrelated samples (294 from the unselected cohort of metastatic patients and 21 from the cohort with RCC hereditary features) were successfully sequenced with a median bait coverage of 883 (min–max: 322–2055; interquartile range [IQR]: 597–1074). Tumor samples had median bait coverage of 397 (min–max: 56–596; IQR: 297–484) and 578 (min–max: 244–725; IQR: 458–628) for the somatic cancer and the hereditary cancer gene panels, respectively.
For the alignment, GRCh37/hg19 assembly was used as reference. HaplotypeCaller and Mutect2 were used for the calling of germline and somatic variants, respectively. Ensembl Variant Effect Predictor annotation tool was used to predict variant impact and only variants with high (nonsense, frameshift, and start/stop loss variants) and moderate (missense variants and inframe indels) impact were considered for the analysis. Germline variants with a minor allele frequency (MAF) > 1% and somatic variants with a MAF > 0.01% in gnomAD were filtered out. Variants with a fraction of altered reads <0.15 were filtered out, except for mosaicism analysis in which all germline variants with a fraction of altered reads <0.30 and with more than ten altered reads were further studied. All low confidence variants were reviewed using the Integrative Genomics Viewer (IGV) and manually curated.
FH germline large-scale exon deletion/duplication events were tested through multiplex ligation-dependent probe amplification (MLPA) Salsa P198-A3 kit (MRC-Holland), following the manufacturer’s instructions, in 30 cases with RCC tumors classified in the anatomopathological reports as papillary (n = 24), mixed features of papillary and other histologies (n = 2), collecting duct (n = 2) or unclassified (n = 2). Fragments were analyzed on ABI 3730xl sequencer (Applied Biosystems) using GeneScan™ 500 LIZ®size standards. Fragment analysis was performed using Peak Scanner Software v1.0 (Thermo Fisher Scientific).
Germline variants classified as pathogenic/likely pathogenic by ClinVar or Leiden Open Variation Database (LOVD) were included in the study. Additionally, loss-of-function (LOF) variants not previously described were classified as likely pathogenic, unless located in the last exon of the gene; in this case they were classified as variants of unknown significance and not considered further.
Immunohistochemistry of FH and 5-hydroxymethylcytosine and tumor methylation assessment through methylation-specific MLPA
Immunohistochemistry (IHC) analyses were performed on 2.5-μm-thick sections from FFPE tumors in an automatic platform (Autostainer Link+, Dako; Discovery XT Ventana, Roche). Briefly, each slide was incubated with mouse monoclonal anti-FH (1:3,500, Santa Cruz Biotechnology, J-13 sc-100743) or rabbit polyclonal anti-5-hydroxymethylcytosine (5hmC) (1:10,000, Active motif, catalog number 39769) after deparaffination and antigen retrieval with the appropriate buffer. This was followed by incubations with the visualization system (EnVision FLEX + Mouse, Dako; OmniMap anti-Rabbit, Ventana, Roche) conjugated with horseradish peroxidase. Immunohistochemical reaction was developed using DAB and nuclei were counterstained with Carazzi’s hematoxylin. FH and 5hmC IHC stainings were evaluated by a pathologist (E.C.).
The methylation status of FH-defective tumors was assessed through methylation-specific (MS)-MLPA following the standard protocol. In brief, 200 ng of DNA were heated for 5 minutes at 98 °C and hybridized for 16 hours at 60 °C with ME042-C1 CIMP probemix (SALSA MS-MLPA Probemix ME042-C1 CIMP, MRC-Holland). Hybridized samples were split in two tubes to perform ligation (undigested reaction) and ligation plus digestion (digested reaction). In the digested reaction tubes, 0.5 µl of HhaI enzyme (SALSA HhaI, MRC-Holland) was added. Ligation was performed at 48 °C during 30 minutes and ligase and HhaI enzymes inactivation was performed by heating at 98 °C for 5 minutes. Ligated probes were amplified and fragments were separated by capillary electrophoresis. Normal kidney FFPE tissue and RCC samples without variants in FH were used as reference.
Statistical analysis
The patients in the unselected cohort of metastatic patients were divided into those carrying a germline variant in a RCC predisposition gene, those carrying a germline variant in other cancer predisposition gene, and those without variants. Categorical variables, including gender, RCC histology, metastatic presentation at diagnosis or in <6 months, and personal and family history of cancers, were compared with the group without variants using chi-squared test or Fisher’s exact test. Continuous variables, such as age at diagnosis, were compared using Mann–Whitney U-test. Differences were considered significant if P values were <0.05.
RESULTS
Characteristics of the patients
Patient demographic and clinical characteristics for the series of 294 metastatic RCC patients and the cohort of 21 RCC patients with clinical features suggestive of hereditary disease are presented in Table 1. For the unselected metastatic series the median age of diagnosis was 60 years with age of onset ranging from 20 to 87 years. The most frequent histologies recorded in the anatomic pathology reports were clear cell (77%) and papillary (9%) RCC. In the series with clinical RCC hereditary features, the median age of diagnosis was 43 years, ranging from 30 to 65 years. Clear cell (67%) and papillary (24%) were the most frequent histologies and 15 (71%) of the patients were metastatic. Six cases (29%) had a personal history in RCC and 4 (19%) a family history in RCC; 6 cases (29%) had bilateral or multifocal RCC. Additional clinical details are provided in Supplementary Table 2.
Germline variants and associated clinical characteristics
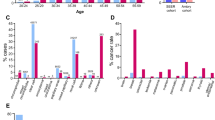
In the unselected metastatic series we identified 4 (1.4%) patients with a variant in a RCC predisposition gene (VHL or FH) and 15 patients with variants in either APC, ATM, BARD1, BRCA1, BRCA2, CHEK2, MSH6, MUTYH, or PMS2 (Fig. 1a, Supplementary Table 3). Variant frequencies among histologic subtypes are shown in Fig. 1b.

(a) Number of patients with a germline variant in RCC predisposition gene (dark gray) or other cancer-related gene (light gray). (b) Percentage of mutated patients depicted by gene type in common RCC histologies: clear cell (ccRCC, n = 226), papillary (pRCC, n = 25) and chromophobe (chRCC, n = 15). The number of mutated patients is expressed in parentheses for each gene.
The patient with the VHL variant had ccRCC at age 40, with no clinical features associated to VHL syndrome either in the patient or in his family, suggesting a de novo event. The three patients with germline FH variants had RCC tumors with papillary type 2 histology, and were diagnosed between 36 and 64 years of age. Two FH-defective patients were females: one with hysterectomy at age 31, and the other with a nephrectomy histopathological report indicating hereditary leiomyomatosis and renal cell cancer (HLRCC)-suggestive histologic features. The remaining patient was a male with cutaneous leiomyomas and had a family history of RCC. Two of the FH cases were metastatic at diagnosis and the other developed metastasis within 4 months. Among the patients with variants in other cancer genes, a BRCA2-mutated male patient had a personal history of prostate cancer and familial history of cancer was present in three additional cases.
When comparing the unselected metastatic patients with and without germline variants (Table 2), the alterations in RCC predisposition genes were more frequent in non-ccRCC versus ccRCC patients (P = 0.0025), this difference being driven by the papillary histology (P = 4.9 × 10−6). The patients with these type of variants were younger (P = 0.036) and they more frequently had a personal and familial history in RCC (P = 6.9 × 10−4 and 0.011, respectively). In contrast, patients with variants in other cancer-related genes had similar clinical characteristics related with renal cancer (e.g., age of RCC diagnosis, RCC histology, personal or familial history in RCC) than those without variants, while a higher prevalence of family history in other cancer types, as expected for these types of genes, was detected (P = 0.020). An increased number of male cases was also observed, maybe reflecting competing risks with female tumors (e.g., in BRCA1, BRCA2).
Among the 21 patients with clinical RCC hereditary features, only 1 patient with a germline pathogenic variant (FH p.R343*) was identified. This was a female patient diagnosed with papillary type 2 RCC, with myomectomy plus hysterectomy at age 36, no known antecedents in RCC, and who developed metastasis 7 months after diagnosis. We searched for low frequency variants in blood and saliva samples from these patients with hereditary clinical features (mean coverage of 692×); however, no evidence for mosaicism in the studied genes was found.
Overall, taking into account the 315 RCC patients included in the study, among 28 cases with non–type 1 papillary RCC histologies (9 papillary type 2, 18 papillary with unspecified subtype, 1 papillary with both type 1 and 2 tumors), 4 patients (14%), all with metastatic disease, harbored a FH germline variant causing HLRCC. This proportion increased to 40% if only papillary type 2 cases were considered.
Tumor secondary point variants, LOH, and global tumor genome hypermethylation
Next-generation sequencing (NGS) was performed in 145 tumor tissues available, including 11 tumors from patients with a pathogenic germline variant (Fig. 2). No somatic second hits were detected in the genes studied. LOH was present in the FH-mutated papillary tumors available and in one case with an ATM variant, while it did not affect BARD1, BRCA2, CHEK2, MSH6, and MUTYH, suggesting that most of these latter alterations do not contribute to RCC development.

For loss of heterozygosity (LOH) analysis (upper panel) tumors were sequenced with the hereditary cancer panel (n = 29 genes). Somatic variants in renal cell carcinoma (RCC) predisposition genes are shown in dark gray and variants in other cancer genes in light gray. For one tumor sample, these sequencing data were not available (in black). Somatic variants in genes commonly mutated in RCC tumors (n = 43 genes analyzed) are presented in the lower panel. High impact variants (loss of function) are shown in dark squares and those with moderate impact (missense and inframe indels) are shown in striped squares. The allele frequencies of the tumor variants are provided inside the squares.
In papillary RCC, the two assessed cases with germline variants in FH had somatic variants in NF2 or ATRX, while the papillary tumors without germline variants had NF2 mutated in one tumor (9%), had no variants in ATRX, but frequent variants in SETD2 (27%), KDM6A (27%), and PBRM1 (18%) (Supplementary Fig. 1). In clear cell histology, the somatic variants found in tumors with and without a germline variants was similar, with VHL, PBRM1, SETD2, and BAP1 being the most frequently mutated genes (44%, 33%, 22%, and 11% in the 9 cases with germline variants versus 69%, 41%, 29%, and 12% in the 112 cases without germline variants; Supplementary Fig. 1). The full list of germline and somatic variants is shown in Supplementary Table 4.
Regarding the molecular characteristics of the FH-deficient RCC tumors, the LOH in FH was confirmed by Sanger sequencing (Fig. 3a) and IHC of FH protein revealed negative staining in only the tumor with a nonsense variant, suggesting that variants in N373 residue inactivate the protein but do not affect its stability (Fig. 3b). The global genome hypermethylation derived from fumarate accumulation was confirmed in FH-mutated tumors through 5hmC IHC staining and MS-MLPA assessment, supporting CpG island methylator phenotype (CIMP) in these tumors (Fig. 3c).

(a) Sanger sequencing chromatograms for FH-positive cases. Sequencing was performed in the blood and in the tumor samples available. (b) Immunohistochemistry (IHC) of FH and 5-hydroxymethylcytosine (5hmC). Representative IHC images obtained for three FH-deficient tumors with tissue available and one pRCC type 2 tumor with no variant in FH (FH-WT). (c) Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) assessing the methylation status of 31 CpG islands in 8 genes, to define CpG island methylator phenotype (CIMP). The tested sample is shown in orange and the reference normal kidney formalin-fixed paraffin-embedded (FFPE) tissue in black. The kit includes methylation-susceptible and reference probes (R).
DISCUSSION
The identification of patients with inherited forms of renal cancer has important clinical implications. For years, 3–5% of renal cancers have been estimated to be familial. However, this estimate derives primarily from early-stage disease research and from studies using low-scale sequencing techniques, hindering large comprehensive genetic screenings in patient populations with specific characteristics. In this regard, a recent study in advanced RCC patients unselected for suspicion of a hereditary syndrome reported that 20% of non-ccRCC histologies carried a germline variant in a cancer susceptibility gene related with RCC or with other tumors, from which half had the potential to direct systemic therapy.10 Thus, in addition to classical clinical hereditary suggestive features, additional characteristics, such as tumor histology or disease stage, may be associated with a higher incidence of hereditary disease.
In our cohort of 294 Spanish unselected patients with metastatic RCC, we found that 0.4% of ccRCC and 6.4% of non-ccRCC cases carried a germline variant in a RCC predisposition gene. A previous study performed on a large series of advanced RCC detected a higher prevalence of germline pathogenic variants in RCC genes in both ccRCC and non-ccRCC cases (1.7% and 12%, respectively).10 This disparity may derive from differences among the cohorts of RCC patients, including the age of the patients (median age of RCC diagnosis was 56 years in Carlo’s study and 60 years in our series), the genetic background (12% of patients with Ashkenazi Jewish ethnicity in Carlo’s cohort) and the proportion of non-ccRCC histological subtypes (31% of non-ccRCC in Carlo’s study versus 16% and in our cohort). Interestingly, in our cohort of patients recruited based on RCC hereditary clinical features, only one patient with a papillary type 2 RCC harbored a germline pathogenic variant. The low mutational incidence observed in this selected cohort suggests that an age of diagnosis ≤46 years without any other additional hereditary suggestive feature (11 of 21 cases in our selected cohort) is a poor criterion to select patients for hereditary RCC screening, especially if they are ccRCC cases. Overall, when combining both unselected and selected cohorts, while the frequency of pathogenic variants was low in ccRCC, 40% of papillary type 2 patients (all with metastatic disease), 14% when considering all non–type 1 papillary cases, carried a FH germline pathogenic variant, supporting that histology and disease stage are important features to consider when selecting patients for genetic screening.
Inherited FH deficiency is known to cause HLRCC, a syndrome that predisposes to aggressive papillary type 2 tumors. In our study in metastatic disease, germline variants in FH were identified in a large proportion of cases with papillary histology. In addition, loss of 5hmC IHC staining and increased genome methylation was observed in FH-mutated tumors, consistent with CIMP. This is in agreement with fumarate accumulation, which inhibits TET activity and results in increased CpG island methylation and decreased 5hmC levels. Type 2 papillary tumors with CIMP are aggressive, highly metastatic, and are associated with a poor survival, differentiating them from other papillary RCC tumors that have better prognosis.15 A recent IHC screening in over 1,000 renal neoplasms obtained through partial/radical nephrectomy and including 400 papillary and 46 unclassified RCCs reported FH deficiency in 0.5% and 4% of these cases, respectively.16 These data are in contrast with the 14% of FH-mutated cases in papillary non–type 1 (or 40% in type 2 papillary RCC) in our series of metastatic cases. Similarly to our study, Carlo et al. found 16% of FH-mutated patients among advanced RCC tumors with papillary or unclassified histology.10 These data support that the prevalence of FH variants is much higher in advanced stage than in localized disease, reflecting the aggressiveness of the FH tumors.
Some histological tumor features, such as prominent nucleoli with perinucleolar halos and multiple architectural patterns within, are suggestive of HLRCC tumors.17 However, the morphologic spectrum of FH-derived RCC is broad and only one of the four FH-mutated patients we reported had HLRCC histopathologic suggestive features annotated. Furthermore, a large study in 114 FH families and 37 associated RCCs found that upon central pathology review by an expert uropathologist, the majority of tumors were papillary type 2, but there were also papillary of unspecified type, and unclassified, tubulocystic, and collecting duct RCC.5 Again, in this series the vast majority of RCCs (82%) were metastatic at diagnosis or rapidly became metastatic.5 Cutaneous leiomyomas, early uterine leiomyomas, multifocal RCC, or a family history in RCC are additional clinical features that may facilitate the identification of individuals harboring a FH variant.18 In our study, two women had hysterectomy at a young age, but only one of our cases had a family history in RCC. In agreement, among seven FH cases Carlo et al. found uterine fibroids in all women, cutaneous leiomyomas in one patient after FH variant detection, and no familial antecedents of RCC. These results point out the difficulty of identifying FH-deficient patients based only on clinical manifestations and suggest that genetic screening would be indicated in all patients presenting with metastatic tumors and papillary type 2 or unconventional RCC histologies. Currently, there is no approved targeted therapy for FH-deficient renal cancers; however, clinical trials in phase 1/2 using targeted therapy for FH are ongoing (ClinicalTrials.gov number: NCT01130519).
The contribution of germline variants in other cancer-associated genes to RCC was also examined, finding that 5% of patients in the metastatic cohort had variants in these genes. MUTYH was the most commonly mutated gene, in all cases with variants in heterozygosis. Biallelic variants in this gene are responsible for MUTYH-associated polyposis syndrome[;19 however, the association of monoallelic variants (which occur in 0.5% of the European population) with malignancies other than colon cancer remains uncertain. Variants in genes with dominant traits were also found, but with the exception of ATM, no LOH was detected in the tumors. Carlo et al. found that 11% of patients carried variants in genes associated to various cancers, but only CHEK2 variant frequency exceeded that of the general population. We could not confirm this result, since in our cohort only one patient carried a CHEK2 variant. In Carlo’s series there was an enrichment in Ashkenazi Jewish patients, who have founder variants such as p.Ser428Phe and p.Ile157Thr in CHEK2 or p.Ile1307Lys in APC; this variability in ethnicity may explain the differences between both studies. To elucidate the contribution of these genes to kidney cancer, further investigation in larger cohorts of patients is needed.
In conclusion, in this study in the metastatic setting a high prevalence of pathogenic germline variants in RCC predisposition genes in non-ccRCC is detected. Taking into account our results and the evidence in the literature, we recommend that all metastatic patients (those with metastasis at diagnosis or those who become metastatic during the course of the disease) with papillary type 2 RCC or unconventional RCC histologies compatible with HLRCC should be offered genetic testing for FH gene. For patients with localized disease and those RCC histologies mentioned, we recommend investigating personal and familial history suggestive of HLRCC (i.e., renal tumors, cutaneous leiomyomas, uterine leiomyomas) and obtaining a tumor review by an expert genitourinary pathologist. For patients with any of these characteristics suggestive of HLRCC, especially but not exclusively in young patients, we also recommend FH genetic screening to search for hereditary disease.
Data availability
Clinical and sequencing data are presented in Supplementary Materials and additional information can be provided upon request.
References
Rini, B. I., Campbell, S. C. & Escudier, B. Renal cell carcinoma. Lancet. 373, 1119–1132 (2009).
Linehan, W. M., Walther, M. M. & Zbar, B. The genetic basis of cancer of the kidney. J. Urol. 170, 2163–2172 (2003).
Lipworth, L., Tarone, R. E. & McLaughlin, J. K. The epidemiology of renal cell carcinoma. J. Urol. 176, 2353–2358 (2006).
Linehan, W. M. The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol. 7, 277–285 (2010).
Muller, M. et al. Reassessing the clinical spectrum associated with hereditary leiomyomatosis and renal cell carcinoma syndrome in French FH mutation carriers. Clin. Genet. 92, 606–615 (2017).
Nguyen, K. A. et al. Advances in the diagnosis of hereditary kidney cancer: Initial results of a multigene panel test. Cancer. 123, 4363–4371 (2017).
Shuch, B. et al. Defining early-onset kidney cancer: Implications for germline and somatic mutation testing and clinical management. J. Clin. Oncol. 32, 431–437 (2014).
Condon, L. T., Ashman, J. N. E., Ell, S. R., Stafford, N. D., Greenman, J. & Cawkwell, L. A population-based familial aggregation analysis indicates genetic contribution in a majority of renal cell carcinomas. Int. J. Cancer 100, 476–479 (2002).
Wu, J. et al. Germline mutations of renal cancer predisposition genes and clinical relevance in Chinese patients with sporadic, early-onset disease. Cancer. 125, 1060–1069 (2019).
Carlo, M. I. et al. Prevalence of germline mutations in cancer susceptibility genes in patients with advanced renal cell carcinoma. JAMA Oncol. 4, 1228–1235 (2018).
Pritchard, C. C. et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N. Engl. J. Med. 375, 443–453 (2016).
Stuttgen, K. et al. Pathogenic germline variants in patients with metastatic breast cancer. JAMA Oncol. 5, 1506–1508 (2019).
Garcia-Donas, J. et al. Single nucleotide polymorphism associations with response and toxic effects in patients with advanced renal-cell carcinoma treated with first-line sunitinib: a multicentre, observational, prospective study. Lancet Oncol. 12, 1143–1150 (2011).
van der Zanden, L. F. M. et al. Description of the EuroTARGET cohort: a European collaborative project on TArgeted therapy in renal cell cancer—GEnetic- and tumor-related biomarkers for response and toxicity. Urol. Oncol. Semin. Orig. Investig. 35, 529.e9–529.e16 (2017).
Linehan, W. M. et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N. Engl. J. Med. 374, 135–145 (2016).
Gupta, S. et al. Incidence of succinate dehydrogenase and fumarate hydratase–deficient renal cell carcinoma based on immunohistochemical screening with SDHA/SDHB and FH/2SC. Hum. Pathol. 91, 114–122 (2019).
Skala, S. L., Dhanasekaran, S. M. & Mehra, R. Hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCC): A contemporary review and practical discussion of the differential diagnosis for HLRCC-associated renal cell carcinoma. Arch. Pathol. Lab. Med. 142, 1202–1215 (2018). https://doi.org/10.5858/arpa.2018-0216-RA
Menko, F. H. et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam. Cancer 13, 637–644 (2014).
Poulsen, M. & Bisgaard, M. MUTYH associated polyposis (MAP). Curr. Genomics 9, 420–435 (2008).
Acknowledgements
This work was supported by the projects RTI2018-095039-B-I00 (Spanish Ministry of Science and Innovation [MCI/AEI], cofunded by the European Regional Development Fund [ERDF]). We thank Dr. Osorio and Dr. Urioste for their work on variant interpretation and Rocío Letón and Fátima Mercadillo for their technical assistance in the MLPA performance. We acknowledge Histopathology Core Unit from the Spanish National Cancer Research Center (CNIO) for their technical support.
Author information
Authors and Affiliations
Contributions
Conceptualization: M.S., C.R.-A. Data curation: M.S., J.L. Formal analysis: M.S., J.L., J.M.R.-R., E.C., C.M.-C., A.C., M.R., C.R.-A. Funding acquisition: C.R.-A. Methodology: M.S., J.L., J.M.R.-R. Resources: M.A.C., G.A., S.H., N.L., L.R., G.d.V., J.G.-D. Supervision: C.R.-A. Writing (original draft): M.S.; C.R.-A. Writing (review and editing): M.S., J.L., J.M.R.-R., E.C., C.M.-C., A.C., M.A.C., G.A., S.H., N.L., M.R., L.R., G.d.V., J.G.-D., C.R.-A.
Corresponding author
Ethics declarations
Ethics Declaration
The project was approved by the institutional review board (IRB) at Instituto de Salud Carlos III (PI 46_2019-v2) and each of the ethical review boards of the participating hospitals (Hospital 12 de Octubre, Hospital Universitario Fundacion Alcorcón, Fundació Althaia-Manresa, Complejo Hospitalario de Navarra, Hospital Universitario HM Sanchinarro, Hospital Clinic i Provincial de Barcelona, Hospital Clinico San Carlos, Complejo Hospitalario de Jaén, Hospital Universitario de Fuenlabrada, Hospital General de Asturias, Hospital Gregorio Marañón, Hospital Infanta Sofía, Hospital Ramón y Cajal, Hospital de la Santa Creu i Sant Pau, Hospital Virgen Rocio, Hospital del Mar, Hospital Universitario Central de Asturias, Hospital Universitario La Paz, Instituto Catalán de Oncología Badalona, Instituto Valenciano de Oncología, Hospital Morales Meseguer, Hospital Universitario Parc Taulí, Hospital Universitario Son Dureta, Hospital Son Llàtzer, Hospital Universitario Virgen de Valme). Written informed consent was obtained from all study patients. The study adheres to the principles set out in the Declaration of Helsinki.
Competing interests
G.A. reports the following competing interests: speakers’ bureau (Kyowa kyrin, Janssen, Bristol Myers Squibb, Roche, Sanofi, Rovi); travel grants (Roche, MSD, Pfizer, Novartis, Astellas, Janssen, Bristol Myers Squibb). G.d.V. reports the following competing interests: receipt of grants/research supports (Ipsen, Pfizer, Roche); receipt of honoraria or consultation fees (Ipsen, Pfizer, Roche, MSD, Merk, Astellas, Jannsen, Novartis, Bayer, BMS). The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Santos, M., Lanillos, J., Roldan-Romero, J.M. et al. Prevalence of pathogenic germline variants in patients with metastatic renal cell carcinoma. Genet Med 23, 698–704 (2021). https://doi.org/10.1038/s41436-020-01062-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-01062-0
