
Abstract
Purpose
Variants in genes encoding sarcomeric proteins are the most common cause of inherited cardiomyopathies. However, the underlying genetic cause remains unknown in many cases. We used exome sequencing to reveal the genetic etiology in patients with recessive familial cardiomyopathy.
Methods
Exome sequencing was carried out in three consanguineous families. Functional assessment of the variants was performed.
Results
Affected individuals presented with hypertrophic or dilated cardiomyopathy of variable severity from infantile- to early adulthood–onset and sudden cardiac death. We identified a homozygous missense substitution (c.170C>A, p.[Ala57Asp]), a homozygous translation stop codon variant (c.106G>T, p.[Glu36Ter]), and a presumable homozygous essential splice acceptor variant (c.482-1G>A, predicted to result in skipping of exon 5). Morpholino knockdown of the MYL3 orthologue in zebrafish, cmlc1, resulted in compromised cardiac function, which could not be rescued by reintroduction of MYL3 carrying either the nonsense c.106G>T or the missense c.170C>A variants. Minigene assay of the c.482-1G>A variant indicated a splicing defect likely resulting in disruption of the EF-hand Ca2+ binding domains.
Conclusions
Our data demonstrate that homozygous MYL3 loss-of-function variants can cause of recessive cardiomyopathy and occurrence of sudden cardiac death, most likely due to impaired or loss of myosin essential light chain function.
Similar content being viewed by others


INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is considered as the leading cause of sudden cardiac death (SCD) among athletes and young adults under the age of 30.1 Dilated cardiomyopathy (DCM) accounts for 30–40% of all heart failure cases and is the leading indication for heart transplantation.2 Variants in genes encoding major components of the sarcomere, the basic contractile unit of striated muscles, are the most common cause of HCM and DCM, which is mostly transmitted in an autosomal dominant fashion.
Autosomal dominant and recessive variants in MYL3, which encodes the ventricular myosin essential light chain (ELC), are a rare cause of HCM with inter- and intrafamilial variability ranging from benign to malignant forms with cardiac failure and SCD.3,4,5,6,7 It has also been suggested that MYL3 variants can result in variable morphological phenotypes.7,8,9 There are, however, no reports of DCM associated with MYL3 variants or cardiomyopathy associated with likely ELC deficiency due to nonsense or essential splice acceptor variants in MYL3.
Here, we describe homozygous MYL3 variants, including likely loss-of-function (LOF) variants, in three unrelated families with recessive HCM or DCM and occurrence of SCD. Pathogenicity of the identified variants is supported by functional assessment of the mutant ELC in zebrafish.
MATERIALS AND METHODS
Patients and genetic analysis
Three unrelated families of Iranian origin were included in this study. Exome sequencing was performed on patient III.7 and individuals III.1 and IV.3 from family A, the proband IV.2 from family B, and individual V.2 from family C, independently. Detailed methods are provided in the Supplementary information. MYL3 transcript NM_000258.2 was used for variant nomenclature.
RESULTS
Clinical characteristics of the patients
Case reports and full descriptions are provided in the Supplementary information. All patients were born to consanguineous parents, either first or second cousins (Fig. 1) presenting with HCM, DCM, or unclassified cardiomyopathy.

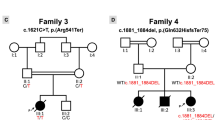
a–c Pedigrees of families A, B, and C. In the pedigree, squares represent males, circles females, black shaded symbols denote cardiomyopathy, open symbols represent clinically asymptomatic individuals, and slash indicates deceased. Family members who were analyzed by next-generation sequencing (arrows) are indicated. Plus (+) and minus (−) signs indicate presence or absence of the MYL3 variants ([+/+] homozygote, [+/−] heterozygote and [−/−] wild-type). d Multiple sequence alignment confirms that the p.(Ala57Asp) substitution affects an evolutionarily conserved residue. e Mutated residues and myosin ELC protein structure: previously identified missense variants with clinical support associated with familial hypertrophic cardiomyopathy (HCM) (black arrows), nearly all located at EF-hand motifs of myosin ELC, and the currently identified variants, p.(Ala57Asp), p.(Glu36Ter) and the splice acceptor variant, c.482-1G>A (red arrows). The actin- and Ca2+-binding sites, proline-rich regions and EF-hand motifs of the myosin ELC are indicated.
Case III.7, a 38-year-old female from family A, was diagnosed with HCM at the age of 26 following dyspnea. Family history was positive for HCM and occurrence of SCD in four family members (Fig. 1a). Case IV.2 is an 8-year-old male from family B (Fig. 1b), who was diagnosed with DCM at the age of 6. He underwent heart transplantation soon after diagnosis. The only other clinical feature noted was horseshoe kidney. The affected brother (IV.1) died at the age of 2 of SCD and renal failure. Case VI.1, from family C, was a female of apparently healthy second cousin parents. She was diagnosed with unclassified cardiomyopathy at the age of 2.5 years and died at the age of 8 years of SCD. Family history was positive for cardiac disease and occurrence of SCD in three family members (V.5, V.6, and V.8), who had been diagnosed with unclassified cardiomyopathy between the ages of 2 and 3 and had died between 5 and 9 years of age (Fig. 1c). There was no record of cardiac evaluation available for these individuals. The 40-year-old mother (V.2) was offered genetic testing following the death of her daughter (VI.1). Unfortunately, there was no tissue available from the deceased individual VI.1.
Affected individuals from the three families showed no evidence of muscle weakness by neurological examination or by history.
Genetic findings
Familial genotyping results and MYL3 variant details are summarized in Fig. 1 and Fig. S2. After filtering of exome sequencing data, no rare, likely pathogenic heterozygous, or homozygous variants in other cardiomyopathy-associated genes were identified. In all families, bidirectional Sanger sequencing analysis validated the identified MYL3 variants confirming a recessive mode of inheritance associated with the cardiomyopathy in families A and B, and in keeping with the consanguineous family history, likely recessive inheritance in family C.
In family A, a homozygous missense variant, c.170C>A (rs139794067), was identified. It leads to substitution of an evolutionarily highly conserved residue (p.[Ala57Asp]) within the EF-hand Ca2+ binding motif of ELC (Fig. 1d, e). In family B, a novel homozygous c.106G>T variant that introduces a premature termination codon at position 36 (p.[Glu36Ter]) was identified (Fig. 1b and S2B). The mother of the proband in family C was found to harbor a heterozygous c.482-1G>A (rs727503300) variant predicted to abolish the acceptor splice-site of MYL3 exon 5 by in silico splicing programs, likely leading to an abnormal or absent protein (Fig. 1c and S2C). Sanger sequencing confirmed the appearance of the c.482-1G>A variant in the father of the proband (Fig. 1c and S2C), suggesting the likelihood of homozygosity of the variant in the deceased proband. The c.170C>A variant was present on one allele in the Genome Aggregation Database (gnomAD) (frequency 0.01588%). The c.106G>T and c.482-1G>A variants were not present in gnomAD or The Greater Middle East (GME) Variome Project. The c.482-1G>A variant was present in the heterozygous state in one individual of our in-house database of 1095 exomes. None of these variants were identified in the homozygous state in any database. The three MYL3 substitutions are suggested to be disease causing by in silico predictors (MutationTaster, PolyPhen-2 [probably damaging], SIFT [deleterious], and PMut [pathogenic]). Based on the American College of Medical Genetics and Genomics (ACMG) criteria,10 the pathogenicity of the MYL3 c.170C>A, c.106G>T, and c.482-1G>A variants were scored as variant of uncertain significance (VUS), pathogenic, and likely pathogenic, respectively (Table S2).
MYL3 variants are unable to rescue zebrafish loss of cmlc1 function
To investigate the functional consequences of the identified variants, we turned to the zebrafish as a model organism. Two cardiac specific myosin light chain orthologs exist in zebrafish, cmlc1 and cmlc2.11 Cmlc1 shows over 70% homology with human ELC, and is highly expressed in zebrafish ventricle and weakly expressed in the atrium, while cmlc2 is homologous to human myosin regulatory light chain (RLC).11 We examined the knockdown phenotype of cmlc1, using a previously characterized morpholino.11 In agreement with published data, we observed cardiac specific defects in cmlc1 morphant embryos (Fig. 2a, b). Compared with the control (Movies S1 and S9), morphants displayed a nonfunctioning heart, characterized by a small ventricle with reduced contractility and a dilated atrium (n = 116/120, p < 0.01, Fig. 2 & Movie S2). Circulating blood is absent to slow in morphants, and ventricular regurgitation is apparent (Movies S2 and S10). To examine conservation of function between human MYL3 and zebrafish cmlc1, RNA for human MYL3 was coinjected with control or cmlc1 morpholino into one-cell staged embryos. Overexpression of MYL3 transcript had no discernible effect on cardiac function in control-injected morphants (Fig.2 and Movie S3). Overexpression of MYL3 RNA in cmlc1 morphants led to a significant improvement in cardiac function, with a notable rescue of heart function (n = 11/29, p < 0.01, Fig. 2c) and significant improvement of ventricular contractions, quantified by measuring the percentage-shortening fraction between diastolic and systolic states (MO:\(\bar x\)7.3, MO + MYL3: \(\bar x\)17.7, P ≤ 0.001, Fig. 2d and Movies S4 and S11). Thus, MYL3 shows conserved function to cmlc1 in the zebrafish and represents a valid system for testing pathogenic function of MYL3 variants.

Knockdown of the zebrafish homolog of MYL3, cmlc1 causes ventricular constriction and atrial dilation that can be rescued by wild-type MYL3 but not mutant RNA harboring the patient variants c.170C>A and c.106G>T. RNA alone or plus morpholino were injected into the cytoplasm of one-cell staged embryos and allowed to develop to 48 hpf. a cmlc1 morphant embryos showed an enlarged atrium (arrow) compared with control (con) morpholino injected embryos. The cmlc1 morphant phenotype was partially rescued by coinjection of c.170C>A RNA; embryos still showed a dilated atrium (arrow), however, displayed some improvement in heart function (categorized by observed blood flow). RNA carrying the c.106G>T variant failed to rescue the cmlc1 morphant phenotype. Scale bar: 200 μm. b Fluorescent antibody stain for atrial specific MYH6 (S46, green) and ventricular enriched MYH1 (MF20, red) at 48 hpf. Note the ventricular rescue in MYL3 coinjected morphants compared with the morpholino injected embryos alone (arrow heads). Scale bar: 10 μm. c Qualitative analysis of cardiac function in embryos injected with either control or cmlc1 morpholino alone, RNA encoding MYL3, MYL3 170C>A, or MYL3 106G>T RNA alone, or coinjected with morpholino and RNA. The heart was considered functional (FH) if both chambers contracted and blood flow was observed. d Quantitative analysis of the ventricular shortening fraction (the percentage area difference between diastolic and systolic state) shows significant improvement of contractility in morphants injected with human MYL3 compared with those injected with variants c.170C>A or c.106G>T (con: \(\bar x\) 35.7±SEM 1.4 n = 10, MO: \(\bar x\)7.3±SEM 0.8 n = 10, MO + MYL3: \(\bar x\)17.7 ± SEM2.3 n = 7, MO + 170C>A: \(\bar x\) 9.0±SEM 1.1 n = 9, MO + 106G>T: \(\bar x\) 6.5 ± SEM0.9 n = 9. ***P ≤ 0.001, ns: not significant). Minigene assay of the MYL3 c.482-1G>A variant. The MYL3 c.482-1G>A variant results in exon 5 skipping or removal of the first nucleotide of exon 5. e Agarose gel of reverse transcription polymerase chain reaction (RT-PCR) products from a minigene assay for the MYL3 c.482-1G>A variant. The wild-type (WT) expression construct produced only products of the expected size (~300 bp), indicative of correct splicing of MYL3 exon 5. Both mutant (MUT) expression clones (generated by site-directed mutagenesis) contain a band around the expected size (~300 bp) and a dominant, smaller band (~220 bp). f Relative abundance of RT-PCR products in each lane calculated by densitometry. g Consensus splice acceptor sequences,19 MYL3 intron 4/exon 5 wild-type and mutant splice acceptor sequences. The variant (red; A) generates a new consensus splice sequence (AG, underlined) shifted one nucleotide compared with wild-type (blue; G). h The ~300-bp RT-PCR product contains a single-nucleotide deletion (G) at the start of MYL3 exon 5 (*), confirming the use of the newly generated splice acceptor sequence. The ~200 bp RT-PCR product shows complete skipping of exon 5. Based on densitometry (f), exon 5 skipping represents the more frequent event. i Amino acid alignment of wild-type myosin ELC protein sequence (Uniprot P08590), and proteins generated with exon 5 deletion (ex5_DEL) or one nucleotide deletion (1nt_DEL). Mismatches and deletions are indicated in red. Both splicing consequences would impact the EF-hand domain 2 (AA 128–163, purple) and EF-hand domain 3 (AA 163–195, green).
To evaluate the functional consequences of the MYL3 variants identified in the patients, we performed rescue experiments using RNAs encoding the missense variant c.170C>A and nonsense variant c.106G>T, both of which convert highly conserved codons in human ELC and in the corresponding zebrafish cmlc1 (Fig. 2a and Movies S5–S8, S12 and S13). Coinjection of c.170C>A RNA with cmlc1 morpholino only partially rescued the morphant phenotype, and embryos still presented with a dilated atrium despite some improvement in cardiac function as observed by more efficient blood circulation, compared with cmlc1 morphants alone (n = 12/26, p < 0.01, Fig. 2a, c and Movies S6 and S12). However, a closer examination of the percentage-shortening fraction showed, despite a slight increase in absolute value, no significant improvement in ventricular contractility (MO: \(\bar x\)7.3, MO + 170C>A: \(\bar x\) 9.0, Fig. 2d). Coinjections of c.170C>A RNA with control morpholino had no effect on heart function (n = 35/36, Fig. 2a, c and Movie S5). The nonsense-coding variant c.106G>T, predicted to activate nonsense-mediated decay of the MYL3 transcript, or if translated to form a nonfunctional truncated protein (159-aa C-terminal deletion), was unable to rescue the cmlc1 morphant phenotype (n = 58/58, Fig. 2a,c, and Movies S7 and S13). Indeed, analysis of ventricular percentage-shortening fraction showed no significant difference in morphants injected with c.106G>T RNA compared with morphants alone (MO: \(\bar x\)7.3, MO + 106G>T: \(\bar x\) 6.5, Fig. 2d). Injection of the c.106G>T RNA in combination with the control morpholino had no effect on zebrafish heart development or function (n = 53/53, Fig. 2a, c and Movie S8). Taken together, these data suggest that both MYL3 variants (p.[Ala57Asp] and p.[Glu36Ter]) are highly likely to be disease causing.
MYL3 c.482-1G>A variant leads to the disruption of EF-hand Ca2+ binding motifs of the myosin ELC
The predominant consequence of the MYL3 c.482-1G>A variant, as assessed using a minigene assay, is skipping of exon 5, which results in a protein change (p.[Gly161_Glu186del]), thereby disrupting the EF-hand Ca2+ binding motifs 2 and 3 (Fig. 2e, f, h, i). The less frequent consequence of this variant is deletion of the first nucleotide of exon 5, causing a frameshift p.(Gly161Valfs*4) (as per Human Genome Variation Society Guidelines [HGVS] guidelines) (Fig. 2g). This would completely abolish the EF-hand 3 domain and disrupt the last 3 AA of the EF-hand 2 domain (Fig. 2i). Detailed results are provided in the Supplementary information.
DISCUSSION
Variants in MYL3 are rarely involved in familial cardiomyopathy,4,5,6,7 and recessive LOF MYL3 variants, thus far, have not been described. The majority of reported variants are, however, associated with SCD at a young age.3,5
Here, we identify recessive variants in MYL3 in three unrelated families presenting with hypertrophic, dilated, or unclassified cardiomyopathy of variable severity from infantile- to early adulthood–onset and SCD. Homozygosity for LOF variants, p.(Glu36Ter) in family B, and likely homozygosity of the splice acceptor variant, c.482-1G>A in family C, appear to cause a more severe phenotype resulting in early SCD and fatality. In addition, we describe a missense variant, p.(Ala57Asp), in a large family with recessive HCM and occurrence of SCD. Lack of clinical features in heterozygous individuals in the families supports the association of cardiomyopathy with homozygosity for the variants. Although the presence of the variants could not be confirmed in family members who died of SCD, homozygosity for the variants is likely due to the consanguinity.
The p.(Ala57Asp) affects a highly conserved amino acid residue in the EF-hand Ca2+ binding motifs near the interface of the myosin heavy and light chains5,7 (Fig. 1d, e). The EF-hand motifs function as calcium sensors, undergoing a conformational change upon binding of calcium that is critical for proper muscle contraction, force production and cardiac function.7 A number of HCM-associated variants, including p.(Glu56Gly) and p.(Ala57Gly), are located in the EF-hand Ca2+ binding motifs5,7 (Fig. 1c). No clinical information is available for the p.(Glu56Gly) variant12 but the p.(Ala57Gly) variant has been reported in two unrelated Korean families and one Japanese patient with dominant familial asymmetric septal hypertrophy and a high incidence of SCD.13 Although heterozygosity for the p.(Ala57Asp) variant has been reported in some cohorts, no clear clinical information or family history of the patients was described to provide support for this variant being pathogenic in the heterozygous state.14,15,16 Homozygosity for the p.(Ala57Asp) has been reported in a single patient with HCM and autosomal recessive mode of inheritance was suggested due to the lack of clinical manifestations in the mother of the proband.17 While induced pluripotent stem cell (iPSC)–derived cardiomyocytes from an asymptomatic individual who was heterozygous for this variant did not show an HCM phenotype,18 this may also reflect the genetic background of the heterozygote and the presence of gene modifiers in this individual.
Our zebrafish data provide support for pathogenicity of the variants tested. Expression of c.106G>T MYL3 or c.170C>A MYL3 in cmlc1 morphants was unable to rescue the characteristic cardiac phenotype, in contrast to wild-type MYL3, which generated embryos with functional ventricles and significant blood flow. This suggests that (1) ELC function is conserved in zebrafish and (2) since the morphant phenotype can be rescued, the morpholino effect is specific to cmlc1, and not a result of potential off-target effects associated with morpholino use. We acknowledge that a proportion of morphant embryos injected with wild-type messenger RNA (mRNA) failed to rescue the heart phenotype; we speculate that this might be due to several limitations imposed by the method. Indeed, microinjection of capped mRNA into the one-cell stage embryo leads to transient expression of gene products that usually degrade by 72 hpf. It is feasible that for some embryos the mRNA has degraded before it has completed its expression effect. In addition, it is likely that uptake of mRNA into the cell is heterogeneous and mosaic. Thus cells destined to a cardiac fate may not always receive the amount necessary for rescue, and this may vary between injected embryos. Another limitation for a technique that relies on injections of coding RNA is the incompatibility for testing the consequences of splicing variants such as c.482-1G>A. However, conducting a minigene assay revealed that the c.482-1G>A splicing variant resulted in a predominant complete deletion of exon 5 of the transcript leading to disruption of the highly conserved EF-hand Ca2+ binding motifs of the myosin ELC.
In conclusion, we identify homozygous variants in MYL3 in three unrelated families with cardiomyopathies and occurrence of SCD, but no skeletal myopathy. The recessive inheritance of the likely LOF MYL3 variants are associated with a particularly severe phenotype resulting in early SCD and lethality. Furthermore, our in vivo zebrafish data provide support for the causality of the nonsense variant c.106G>T and missense variant c.170C>A, and that perturbations in myosin ELC function lead to cardiac specific defects.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary material.
References
Marian, A. J. & Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ Res 121, 749–770 (2017).
Haas, J. et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J 36, 1123–35a (2015).
Kazmierczak, K. et al. Discrete effects of A57G-myosin essential light chain mutation associated with familial hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol 305, H575–H589 (2013).
Kabaeva, Z. T. et al. Systematic analysis of the regulatory and essential myosin light chain genes: genetic variants and mutations in hypertrophic cardiomyopathy. Eur J Hum Genet 10, 741–748 (2002).
Hernandez, O. M., Jones, M., Guzman, G. & Szczesna-Cordary, D. Myosin essential light chain in health and disease. Am J Physiol Heart Circ Physiol 292, H1643–H1654 (2007).
Poetter, K. et al. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet 13, 63–69 (1996).
Yadav, S., Sitbon, Y. H., Kazmierczak, K. & Szczesna-Cordary, D. Hereditary heart disease: pathophysiology, clinical presentation, and animal models of HCM, RCM, and DCM associated with mutations in cardiac myosin light chains. Pflugers Arch 471, 683–699 (2019).
Olson, T. M., Karst, M. L., Whitby, F. G. & Driscoll, D. J. Myosin light chain mutation causes autosomal recessive cardiomyopathy with mid-cavitary hypertrophy and restrictive physiology. Circulation. 105, 2337–2340 (2002).
Caleshu, C. et al. Furthering the link between the sarcomere and primary cardiomyopathies: restrictive cardiomyopathy associated with multiple mutations in genes previously associated with hypertrophic or dilated cardiomyopathy. Am J Med Genet A 155A, 2229–2235 (2011).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424 (2015).
Chen, Z. et al. Depletion of zebrafish essential and regulatory myosin light chains reduces cardiac function through distinct mechanisms. Cardiovasc Res 79, 97–108 (2008).
Richard, P. et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 107, 2227–2232 (2003).
Lee, W. et al. Different expressivity of a ventricular essential myosin light chain gene Ala57Gly mutation in familial hypertrophic cardiomyopathy. Am Heart J 141, 184–189 (2001).
Fokstuen, S. et al. Rapid detection of genetic variants in hypertrophic cardiomyopathy by custom DNA resequencing array in clinical practice. J Med Genet 48, 572–576 (2011).
Berge, K. E. & Leren, T. P. Genetics of hypertrophic cardiomyopathy in Norway. Clin Genet 86, 355–360 (2014).
Rubattu S., et al. A Next-Generation Sequencing Approach to Identify Gene Mutations in Early- and Late-Onset Hypertrophic Cardiomyopathy Patients of an Italian Cohort. Int J Mol Sci. 2016; 17.
Jaafar, N. et al. Spectrum of Mutations in Hypertrophic Cardiomyopathy Genes Among Tunisian Patients. Genet Test Mol Biomarkers 20, 674–679 (2016).
Ma, N. et al. Determining the Pathogenicity of a Genomic Variant of Uncertain Significance Using CRISPR/Cas9 and Human-Induced Pluripotent Stem Cells. Circulation. 138, 2666–2681 (2018).
Bryen, S. J. et al. Pathogenic Abnormal Splicing Due to Intronic Deletions that Induce Biophysical Space Constraint for Spliceosome Assembly. Am J Hum Genet 105, 573–587 (2019).
Acknowledgements
We thank the family members who provided samples and clinical information for this study. The study was supported by grants from the European Union’s Seventh Framework Programme for research, technological development and demonstration under grant agreement number 608473 (H.T.) and the Swedish Research Council (H.T.). N.L. was supported by Australian National Health and Medical Research Council (NHMRC) Principal Research Fellowship (APP1117510), N.L. and H.G. by NHMRC EU Collaborative grant APP1055295. The funders had no role in the design of the study and collection, analysis, decision to publish, interpretation of data, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization: Y.J., H.T.; Data curation: D.P.S.O., L.E., J.C., M.T.T., R. M., M.Y., M.L.E.G., H.G., G.S., N.M., A.S.; Formal Analysis: D.P.S.O., L.E., J.C., A.Y.B.W., H.G.; Funding acquisition: Y.J., N.L., H.T.; Investigation: D.P.S.O., J.C., Y.J., H.T.; Resources: M.T.T., C.H., N.M., Y.J., H.T.; Writing – original draft: Y.J., H.T.; Writing – review & editing: N.L., Y.J., H.T.
Corresponding authors
Ethics declarations
COMPETING INTERESTS
The authors declare no competing interests.
ETHICS DECLARATION
Written informed consent for genetic testing and publication of relevant findings was obtained from all patients or their parents. The study was conducted in line with the Declaration of Helsinki and was approved by the relevant institutional review boards in Iran (Shahid Chamran University of Ahvaz), Sweden (Ethics Review Committee in the Gothenburg Region), Australia (Human Research Ethics Committee of the University of Western Australia), and England (St George’s University of London). All zebrafish studies were conducted in compliance with the UK Animals (Scientific Procedures) Act 1986. The studies were approved by a local (St George’s University of London) ethical review committee and licensed under the Home Office granted project license P3DFD3131.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Osborn, D.P.S., Emrahi, L., Clayton, J. et al. Autosomal recessive cardiomyopathy and sudden cardiac death associated with variants in MYL3. Genet Med 23, 787–792 (2021). https://doi.org/10.1038/s41436-020-01028-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-01028-2
This article is cited by
-
Familial dilated cardiomyopathy in a child: a case report
BMC Pediatrics (2024)
-
Investigating cardiac genetic background in sudden infant death syndrome (SIDS)
International Journal of Legal Medicine (2024)
-
The Genetic Factors Influencing Cardiomyopathies and Heart Failure across the Allele Frequency Spectrum
Journal of Cardiovascular Translational Research (2024)
-
EMQN: Recommendations for genetic testing in inherited cardiomyopathies and arrhythmias
European Journal of Human Genetics (2023)
-
Proteomic profiling of sudden cardiac death with acquired cardiac hypertrophy
International Journal of Legal Medicine (2023)
