
Abstract
Purpose
In this study we investigate the disease etiology in 12 patients with de novo variants in FAR1 all resulting in an amino acid change at position 480 (p.Arg480Cys/His/Leu).
Methods
Following next-generation sequencing and clinical phenotyping, functional characterization was performed in patients’ fibroblasts using FAR1 enzyme analysis, FAR1 immunoblotting/immunofluorescence, and lipidomics.
Results
All patients had spastic paraparesis and bilateral congenital/juvenile cataracts, in most combined with speech and gross motor developmental delay and truncal hypotonia. FAR1 deficiency caused by biallelic variants results in defective ether lipid synthesis and plasmalogen deficiency. In contrast, patients’ fibroblasts with the de novo FAR1 variants showed elevated plasmalogen levels. Further functional studies in fibroblasts showed that these variants cause a disruption of the plasmalogen-dependent feedback regulation of FAR1 protein levels leading to uncontrolled ether lipid production.
Conclusion
Heterozygous de novo variants affecting the Arg480 residue of FAR1 lead to an autosomal dominant disorder with a different disease mechanism than that of recessive FAR1 deficiency and a diametrically opposed biochemical phenotype. Our findings show that for patients with spastic paraparesis and bilateral cataracts, FAR1 should be considered as a candidate gene and added to gene panels for hereditary spastic paraplegia, cerebral palsy, and juvenile cataracts.
Similar content being viewed by others

INTRODUCTION
Ether lipids constitute a special class of lipids characterized by the presence of an ether bond at the sn-1 position of the glycerol backbone instead of an ester bond that is found in most other lipids (Fig. 1a). Ether lipids can be subdivided into neutral lipids and phospholipids. The neutral ether lipids are poorly characterized and their functions are largely unknown. Ether phospholipids (Fig. 1), which include plasmalogens and account for ∼20% of the phospholipids in humans, are present in cellular membranes of most tissues, with particularly high levels in the central nervous system, heart, kidney, and white blood cells.1,2 Plasmalogens are believed to play a role in membrane dynamics, as antioxidants and/or as signaling molecules.3 They have been implicated in the pathology of several neurodegenerative disorders, such as Alzheimer disease and Parkinson disease,1,2 but their importance is demonstrated unequivocally by the existence of various inherited disorders with a severe multisystem phenotype in which the biosynthesis of ether lipids is impaired.1,2,4 These disorders include five different types of rhizomelic chondrodysplasia punctata (RCDP), with a primary defect in plasmalogen synthesis, and the Zellweger spectrum disorders (ZSD), with a secondary plasmalogen deficiency due to defective biogenesis of peroxisomes.

a Molecular structures of glycerolipids and glycerophospholipids. These types of lipids contain a glycerol backbone (red) attached to one or two fatty acid/fatty alcohol (blue) substituents at the sn-1 and sn-2 position and a headgroup R (green) attached at the sn-3 position. When R is a hydrogen or a fatty acid the molecule is a glycerolipid; when R is a phosphate linked to a species characteristic headgroup (choline, ethanolamine, etc.) the molecule is a glycerophospholipid, also known as a phospholipid. In ether phospholipids, the fatty linkage at the sn-1 position is either an ether (plasmanyl) or a vinyl ether (plasmenyl); the latter substitution defines the subclass that is known as plasmalogens. In analogy to ether phospholipids, ether variants of glycerolipids also exist with an ether fatty linkage on the sn-1 position. b Ether lipid biosynthesis. Dihydroxyacetone phosphate (DHAP)-acyltransferase (DHAPAT), esterifies DHAP at the sn-1 position with a long-chain fatty acyl-CoA yielding 1-acyl-DHAP. Next, alkylglycerone phosphate synthase (AGPS) replaces the acyl unit by a fatty alcohol, produced by reduction of an acyl-CoA by fatty acyl-CoA reductase 1 (FAR1), resulting in the formation of 1-alkyl-DHAP. 1-Alkyl-DHAP is then reduced to 1-alkylglycerolphosphate by acyl/alkyl-DHAP reductase (PexRAP), which is converted to 1-alkyl-2-acylglycerol (DG[O]) at the endoplasmic reticulum (ER). Exogenous 1-alkylglycerol (for example 1-hexadecylglycerol [HDG]) can be phosphorylated to 1-alkylglycerolphosphate, which then bypasses the peroxisomal steps in ether lipid biosynthesis. DG[O] can be converted to plasmanyl-PE and plasmanyl-PC by condensation with CDP-ethanolamine and CDP-choline, respectively. Creation of a vinyl ether by desaturation in the ER yields plasmenyl-PE, which can be converted to plasmenyl-PC, both of which are called plasmalogens. PC and PE ether phospholipids, thus including plasmanyl and plasmenyl-species, are collectively called PC[O] and PE[O], respectively. DG[O] can also be acylated which yields 1-alkyl-2,3-diacylglycerol (TG[O]).
The biosynthesis of ether (phospho)lipids is a multistep process involving different enzymes; the first three steps take place in the peroxisome and the subsequent steps at the endoplasmic reticulum (Fig. 1b).3 After peroxisomal synthesis of 1-acyl-dihydroxyacetone phosphate (1-acyl-DHAP), the acyl unit is replaced by a fatty alcohol resulting in the formation of 1-alkyl-dihydroxyacetone phosphate (1-alkyl-DHAP). The fatty alcohols for this reaction are produced by peroxisomal fatty acyl-CoA reductase 1 (FAR1), which accepts both saturated and (mono-)unsaturated fatty acyl-CoAs of 16 and 18 carbon atoms as substrates and reduces them to the corresponding fatty alcohols using NADPH as cofactor.5,6 1-Alkyl-DHAP is then reduced to 1-alkylglycerolphosphate in the peroxisome and further converted to ether (phospho)lipids in several steps at the endoplasmic reticulum (Fig. 1b).
FAR1 is localized in the peroxisomal membrane and believed to be the rate-limiting enzyme in ether (phospho)lipid biosynthesis. FAR1 is a tail-anchored protein of 515 amino acids with an N-terminal domain corresponding to amino acids 1–465 facing the cytosol, a transmembrane domain corresponding to amino acids 466–483 and a C-terminal domain corresponding to amino acids 484–515 located in the peroxisomal matrix. The region between amino acids 451–507 is required for the peroxisomal targeting of FAR1 that depends upon Pex19p.7 The activity of FAR1 is regulated at the protein level by active protein degradation in response to increased levels of plasmalogens.6 Accordingly, in cell lines with a plasmalogen deficiency, FAR1 protein levels and activity are elevated, while exogenous supplementation of plasmalogens (or precursors thereof) to these cells results in normalization of FAR1 protein levels. The sensing of plasmalogen levels in this feedback regulatory mechanism is believed to take place in the inner leaflet of the plasma membrane,6 but the precise mechanism remains enigmatic.
FAR1 deficiency (also called RCDP type 4, MIM 616154) has been described in very few individuals.8,9 The autosomal recessive disorder results in intellectual disability, congenital cataracts, growth retardation, and epilepsy. Rhizomelia and skeletal changes, seen in other types of RCDP (types 1–3 caused by defects in PEX7 [MIM 215100], dihydroxyacetone phosphate-acyltransferase [DHAPAT] [MIM 222765], and alkylglycerone phosphate synthase [AGPS] [MIM 600121], respectively), are absent. Similar to the other RCDP types, FAR1-deficient patients have a deficiency of plasmalogens.8
We now report a cohort of patients with an autosomal dominant disorder with spastic paraparesis and early-onset cataract caused by three different heterozygous de novo variants in FAR1. All variants are located in the same codon and change the arginine at amino acid position 480 into a cysteine, histidine, or a leucine. Functional analysis revealed that these variants do not result in a FAR1 deficiency but instead in the loss of feedback regulation by cellular plasmalogen levels, leading to increased plasmalogen levels, increased FAR1 protein levels, and profound lipidomic changes in patient-derived cells.
MATERIALS AND METHODS
Clinical, laboratory evaluation and genetic testing of patients
See Table 1 for clinical features and Supplementary information 1 for case reports and detailed genetic analyses. Clinical trio (patients 2–10 and 12) or quartet (patient 1, with unaffected sibling) exome analysis was performed for patients 1–10 and 12 and an exome-based cerebral palsy panel was performed for patient 11.
Fibroblast culture, 1-O-hexadecyl-sn-glycerol and C17:0 fatty acid incubations
Primary skin fibroblast cell lines from six anonymized control subjects and patients 1–3 were cultured in HAM F-10 medium supplemented with 10% fetal calf serum (HAM/10% FCS, Invitrogen, Carlsbad, CA), 25 mM HEPES, 100 U/mL penicillin, 100 µg/mL streptomycin, and 250 µg/mL amphotericin in a humidified atmosphere of 5% CO2 at 37 °C. For 1-O-hexadecyl-sn-glycerol (HDG) loading, patient fibroblasts were incubated for 24 or 48 hours with HDG (Sigma, St. Louis, MO) dissolved in ethanol and diluted to 10 µM in HAM/10% FCS medium. For the 48-hour incubation period medium containing 10 µM HDG was refreshed after 24 hours. For the C17:0-acid labeling experiments fibroblasts were incubated for 72 hours with heptadecanoic acid (C17:0) dissolved in dimethyl sulfoxide and diluted to 40 µM in HAM/10% FCS medium. Cells were harvested by trypsinization (0.5% trypsin-EDTA, Invitrogen), and washed with phosphate-buffered saline and twice with 0.9% NaCl, followed by centrifugation at 4 °C (523g for 5 minutes) to obtain cell pellets, which were stored at −80 °C.
Plasmalogen measurement in fibroblasts
Fibroblast pellets were homogenized in phosphate-buffered saline by sonication and 200 μg of protein was used to measure plasmalogen levels using gas chromatography as described previously.10 This method is based on acidic methanolysis of lipids, converting 1-O-(alkenyl)-groups to dimethylacetals and fatty acids to methyl esters. The relative amount of plasmalogens is calculated as C16:0- and C18:0-dimethylacetals as percentage of their corresponding fatty acid methyl ester.
FAR1 enzyme measurement in fibroblasts using gas chromatography–mass spectrometry (GC-MS) analysis
Fibroblast pellets were resuspended in 0.9% NaCl and sonicated twice at 40 W. The assay had a final volume of 100 µl of 100 mM TRIS buffer, pH 7.4, 10 mM MgCl2, 0.12 mg BSA, 0.1 mM NADPH, 1 mM glucose-6-phosphate, 1U glucose-6-phosphate dehydrogenase (G6PDH) (NADPH regenerating system), 25 µM heptadecanoyl-CoA (C17-CoA), and 40 µg of fibroblast homogenate protein. Addition of the homogenate started the reaction, which was allowed to proceed for 2 hours at 37 °C in a shaking water bath. Four hundred microliters of methanol/chloroform 75/25 plus 400 pmol of C19-alcohol (C19OH) was added while vortexing to terminate the reaction. Protein precipitates were removed by centrifugation (5 minutes at 18,000g) at 4 °C and supernatants were taken to dryness under a nitrogen stream. Fatty alcohols were silylated to their corresponding tert-butyldimethylsilyl (TBDMS) derivate using 60 µl MTBSTFA at 80 °C for 30 minutes. One µl of this mixture was injected onto an Agilent 7890/5977 GC-MS equipped with a CPsil-19 capillary column (25 m*0.25 mm ID, film thickness 0.20 µm) using helium as carrier gas. MS acquisition was performed in the SIM mode, monitoring the characteristic [M-57]+ fragment of molecular ions of C17-alcohol (C17OH)-TBDMS; m/z 313 and C19OH-TBDMS; m/z 341. Quantification was done using Masshunter software using a C17OH calibration curve.
Immunoblot and immunofluorescence analysis
Immunoblotting of fibroblast homogenates from control subjects and patients 1–3 was performed essentially as described previously.11 Membranes were blocked with Intercept® (PBS) Blocking Buffer and rabbit polyclonal antibodies against FAR1 were used (Sigma, HPA017322, 1:250). As a loading control, the membranes were reprobed with a monoclonal antibody against β-actin (Sigma, A5441, 1:10.000). Immunofluorescence microscopy was performed essentially as described12 with an additional guanidine HCl treatment13 using anti-FAR1 (1:400) and goat antirabbit Alexa Fluor 555 (Invitrogen, A21429, 1:200). Peroxisomal membranes were labeled with anti-ABCD3 (Sigma, SAB4200181, 1:400) and biotinylated α-mouse antibodies (DAKO, E433, 1:200), and streptavidine-FITC (Thermo Fisher, 11-4317-87, 1:200). Slides were examined in the Leica TCS-SP8 filter-free spectral confocal microscope (63× magnification) and processed with the Leica Application Suite AF Lite software (Leica).
Lipidomics
Lipidomics in fibroblasts, also in the C17:0 fatty acid labeling experiment, was performed as described previously.14
RESULTS
Genetic analysis
We identified 12 individuals from around the world with heterozygous de novo missense variants in FAR1 (NM_032228.6): 4 patients with variant c.1439G>A (p.Arg480His), 7 patients with variant c.1438C>T (p.Arg480Cys), and 1 patient with variant c.1439G>T (p. Arg480Leu) (Table 1). All three variants were not reported in the gnomAD database and all are predicted to be deleterious by various pathogenicity prediction programs. Remarkably, all de novo variants are located in the same codon, p.Arg480, which is located in the predicted transmembrane region of FAR1, formed by amino acids 466–483.
Clinical features of patients with heterozygous FAR1 p.Arg480Cys/His/Leu variants
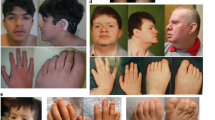
The clinical details of the 12 patients are summarized in Table 1 and are detailed in Supplementary information 1. The patients were between 2 and 19 years of age at the time of data collection but all exhibited neurological symptoms in their first years of life. They presented with pyramidal tract dysfunction, exhibiting spastic di- or paraparesis (12/12), lower limb hypertonia, truncal hypotonia (6/12), and (ankle) clonus. In addition, all patients had bilateral cataracts (12/12), which were congenital in 5/12 and acquired in the other 7/12. Seizures occurred in 8/12 patients, mainly in the first months of life, and appeared amenable to therapy with barbiturates, levetiracetam, and/or oxcarbazepine. Antiepileptic treatment could be discontinued without recurrence of seizures in 4/8 patients. Cognitive delay and intellectual disability were less common in the cohort (3/12). Speech development was delayed in 10/12 patients, with receptive speech outperforming expressive speech in 3 patients. Growth delay was noted in just one patient. There were no dysmorphic features; two patients presented with macrocephaly. Brain imaging was within normal limits in 10/12 patients; one patient had abnormal temporal lobe morphology and ventricular prominence, but white matter was normal. One patient had benign enlargement of subarachnoid spaces.
Functional consequences of the FAR1 variants at the biochemical, protein, and enzyme level
We studied the effect of the identified FAR1 variants in fibroblasts of patients 1–3 (harboring the p.Arg480His [n = 2] and p.Arg480Cys variants [n = 1]). Because of the reported role of FAR1 in the regulation of plasmalogen biosynthesis we first measured plasmalogen levels. In contrast to previously reported patients with FAR1 deficiency, the plasmalogen levels in fibroblasts of the patients were not decreased, but increased (see Fig. 2a). Furthermore, the enzyme activity of FAR1 was markedly increased in the fibroblasts of patients 1–3 (379, 323, and 310 pmol/[h.mg protein], respectively) compared with the FAR1 activity in fibroblasts of control subjects (74, 90, 101 pmol/[h.mg protein]) (Fig. 2b). Moreover, immunoblot analysis showed clearly increased FAR1 protein levels in the patients’ fibroblasts (Fig. 2c). Immunofluorescence microscopy analysis showed that FAR1 is normally localized to peroxisomes in patients’ fibroblasts (see Fig. 2d for representative image) showing that p.Arg480His/Cys variants do not interfere with peroxisomal targeting of FAR1. Also, peroxisomes were of normal size and shape and peroxisomal functions (β-oxidation of very long chain fatty acids and activity of DHAPAT) were normal (not shown). We were not able to study the functional effects of the p.Arg480Leu variant because no fibroblasts were available from the only patient with this genotype.

a Plasmalogen levels in cultured skin fibroblasts from a reference population (Ref), controls (C) (Ref and C, both black diamonds) and FAR1 p.Arg480His/Cys patients (gray, P1 = square, P2 = circle, P3 = triangle). The control samples were analyzed in the same experiments as the patient samples. C16:0- and C18:0-plasmalogens are expressed as % of total palmitate (C16:0) and stearate (C18:0), respectively, and were significantly elevated when compared with the control samples (t-test, two sided, p < 0.0001, indicated by **). b FAR1 enzyme activity in cultured skin fibroblasts of controls (C1–C3, black) and patients (P1–P3, gray). Measurements were performed in duplicate (except for C2). c Immunoblot analysis of FAR1 in fibroblast homogenates of controls and patients. The upper panel shows the (cropped) immunoblot using antibodies against FAR1 (59 kDa) and the lower panel the same blot reprobed with antibodies against β-actin as loading control. d Immunofluorescence microscopy analysis in fibroblasts of P2 of peroxisomal membrane protein ABCD3 (green, upper left) and FAR1 (red, upper right) and overlay (lower left) and magnification (lower right). FAR1 p.Arg480His/Cys variants are normally localized to peroxisomes.
In summary, both the p.Arg480His and the p.Arg480Cys variants result in increased FAR1 protein levels, increased FAR1 enzyme activity and increased plasmalogen levels.
FAR1 variants p.Arg480His/Cys compromise modulation of FAR1 levels by cellular plasmalogen levels
Since it has been demonstrated that, under normal conditions, FAR1 levels and enzyme activity are negatively regulated by cellular plasmalogen levels, we hypothesized that the p.Arg480 variants compromised this negative feedback mechanism in the patients. To investigate the plasmalogen-dependent regulation of FAR1 levels we incubated patient and control fibroblasts with 10 μM 1-O-hexadecyl-sn-glycerol (HDG) for 24 and 48 hours and studied the effect on FAR1 protein level. HDG can be phosphorylated to 1-O-hexadecyl-sn-glycerolphosphate, which is an ether lipid synthesis intermediate, downstream of FAR1 (Fig. 1b). HDG thus bypasses the peroxisomal steps and leads to increased plasmalogen levels, which, through feedback regulation, should result in decreased levels of FAR1.6 In control fibroblasts, HDG treatment increased C16:0-plasmalogen levels 199–291% (mean ± SD: 249 ± 31%) (Fig. 3c) and resulted in a 31–47% decrease (mean ± SD: 40 ± 7%) in FAR1 protein levels (Fig. 3a, b). In the patients’ cells, treatment with HDG also increased C16:0-plasmalogen levels 130-224% (mean ± SD: 160 ± 35%) (Fig. 3c), but this did not result in a consistent decrease of FAR1 protein levels (mean ± SD: 101 ± 17%) (Fig. 3a, b).

Fibroblasts of control subjects (n = 6) and p.Arg480His/Cys FAR1 patients (P1–P3) were treated for 24 and 48 hours with 10 μM HDG followed by immunoblot analysis for FAR1 and measurement of the plasmalogen levels. Two independent experiments were performed (a indicating the first experiment, b the second), each including three (×2) control cell lines and the three patient cell lines (P1 = square, P2 = circle, P3 = triangle). a Immunoblot analysis of FAR1 in fibroblast homogenates of controls and patients using antibodies against FAR1 (59 kDa) and β-actin as loading control (same blot reprobed). Blots (cropped) of both experiments are shown. b Quantification of the FAR1 protein level as ratio over β-actin in control and patient cells in untreated and HDG-treated cells. Results are shown for each cell line as % of the untreated condition. c C16:0-Plasmalogen levels (expressed as % of total palmitate [C16:0]) in untreated and HDG-treated control and patient cells.
It should be noted that in untreated patients’ cells the mean C16:0-plasmalogen levels were already twofold higher than the mean levels in the six control lines and almost as high as the C16:0-plasmalogen levels of HDG-treated controls (Fig. 3c). Despite these high plasmalogen levels, FAR1 protein levels in the patients’ cells were threefold higher than in the HDG-treated controls (Supplementary information 2).
Ether lipid synthesis is increased in patients with p.Arg480His/Cys FAR1 variants
To investigate whether ether lipid synthesis is increased we incubated fibroblasts with C17:0 fatty acid (C17:0-acid). This fatty acid is normally present in low levels and when incorporated into complex lipids is therefore easily tracked by analyzing odd-numbered lipid species that are synthesized from it. In addition to incorporation of C17:0-acid in nonether lipids, allowing monitoring of nonether lipid synthesis, FAR1 converts C17:0-acid into C17:0-alcohol, which is used for the synthesis of ether lipids, providing a way to track ether lipid synthesis (Fig. 4f). Lipidomic analysis of LPC(17:0) and LPC(O-17:0) was used to compare nonether and ether lipid synthesis, respectively, in fibroblasts of controls and patients. Incorporation of C17:0-acid in LPC(17:0) was similar in controls and patients whereas FAR1-dependent incorporation of C17-alcohol in LPC(O-17:0) was almost fourfold higher in patients than in controls (Fig. 4g) confirming that ether lipid synthesis indeed is elevated in patients with p.Arg480His/Cys variants.

Summation of lipidomic species per major class for controls (n = 3, black diamonds), patients 1, 2, and 3, (P1 = square, P2 = circle, P3 = triangle); mean is shown and x-fold difference of the patient mean compared with that of controls if p ≤ 0.005. a Diradylphospholipid species, b monoradylphospholipid species, and c neutral lipid species (“radyl” means either a fatty acid or fatty alcohol substituent). DG diacylglycerol, DG[O] 1-alkyl-2-acylglycerol, (L)PC (lyso)phosphatidylcholine, (L)PC[O] (lyso)plasm(a/e)nylcholine, (L)PE (lyso)phosphatidylethanolamine, (L)PE[O] (lyso)plasm(a/e)nylethanolamine, TG triacylglycerol, TG[O] 1-alkyl-2,3-diacylglycerols. d LPC[O] levels 16:0–18:0 are elevated in patients with the FAR1 variants (10log-scale). e 2Log(fold-change) of the average levels of patients and controls for PC[O] and PC species with 40–44 carbon atoms in the fatty acid side chains. PC[O] species with more double bonds (containing polyunsaturated fatty acids) accumulated more in patients whereas the corresponding PC species were reduced. f Overview of the experiment to assess ether lipid and nonether lipid synthesis from exogenous C17:0-acid in control fibroblasts and patient fibroblasts with the p.Arg480His/Cys FAR1 variants. C17:0-acid can be directly incorporated in nonether lipids or converted by FAR1 to a C17:0-alcohol which in turn can be incorporated in ether lipids. g Comparison of incorporation of C17:0-acid and C17:0-alcohol in LPC(17:0) and LPC(O-17:0), respectively. C17:0-acid is incorporated in comparable amounts in LPC(17:0) in both controls and patients whereas C17:0-alcohol incorporation in LPC(O-17:0) is fourfold elevated in patients when compared with control.
Profound changes in the fibroblast lipidome of patients with p.Arg480His/Cys FAR1 variants
To investigate the consequences of the compromised modulation of FAR1 levels on the lipidome, we performed lipidomics in fibroblasts of patients 1–3 and compared this with three control cell lines. We detected a significant accumulation of different ether lipid species, in particular plasm(a/e)nylcholine ether phospholipid species (PC[O]) and neutral ether lipid species including 1-alkyl-2,3-diacylglycerols (TG[O], the ether lipid equivalents of triacylglycerols [TG]), and 1-alkyl-2-acylglycerols (DG[O], the ether lipid equivalents of diacylglycerols [DG]) (Fig. 4a–c). There was a trend toward increased levels of (lyso)plasm(a/e)nylethanolamine ether phospholipid species (PE[O] and LPE[O]) but this did not reach statistical significance due to variation in the different patients’ cells (Fig. 4a, b). For the nonether lipids, DG levels appeared lower and total TG levels showed a trend to higher values in patients (both not significant). Total levels of the major phospholipids phosphatidylcholine (PC) and phosphatidylethanolamine (PE) were both significantly decreased, with PE levels drastically reduced (Fig. 4a, b). Despite these large differences, the total amount of choline- and ethanolamine-containing lipids (PE + PE[O] and PC + PC[O]), both relative and absolute, remained quite constant (Supplementary fig. S1). Further examination of the LPC[O] species, which contain only a fatty alcohol on the sn-1 position, showed that species with a C16:0-, C17:0-, or C18:0-alcohol were the most elevated species (Fig. 4d), whereas other LPC[O] species with longer fatty alcohol substituents were less elevated, or not affected (Supplementary fig. S2a). These LPC[O] species thus reflect the substrate specificity of FAR1, which preferably uses C16–C18 fatty alcohols.5 Although virtually all PC[O] species were elevated in the patients’ cells, those with 40–44 carbon atoms in the fatty acid side chains (summation of the carbon atoms in both side chains) with higher numbers of double bonds (and thus containing polyunsaturated fatty acids [PUFAs]) showed the highest fold changes. In contrast, the levels of the corresponding PUFA-containing PC species were reduced in the patients’ cells (Fig. 4e). Similar redistribution of PUFA-containing species was also seen in cardiolipin, phosphatidylserine (deficiency), and TG (accumulation) (Supplementary fig. S2b–d). In summary, the elevated FAR1 activity results in a considerably increased synthesis of ether (phospho)lipids together with a concomitant reduction of corresponding nonether species and affects the distribution of PUFAs among the different main lipid classes.
DISCUSSION
FAR1 loss-of-function versus heterozygous de novo FAR1 variants
In this paper we describe a cohort of 12 patients with different de novo missense variants in the FAR1 gene, but all resulting in an amino acid change at position 480 (p.Arg480Cys/His/Leu). The first indication for this cohort came from clinical exome analysts at GeneDx who noticed a trend in the occurrence of heterozygous variants in FAR1 involving the p.Arg480 of the FAR1 protein. This was internally explored in a cohort of 42,983 exome trios and 9205 exome-based specific gene list trios that included FAR1 analysis. This led to the identification of eight cases. Submission of the variants to the GeneMatcher platform15 resulted in the identification of additional cases and initiated an international collaboration to functionally characterize the p.Arg480 variants in FAR1. All patients have significant neurological symptoms including spastic paraparesis with truncal hypotonia, mild to moderate speech and gross motor developmental delay, combined with bilateral congenital or juvenile cataracts and early-onset epilepsy in most. Functional analyses, including fibroblast plasmalogen measurements, FAR1 enzyme activity assays, and lipidomics showed that the biosynthetic function and peroxisomal localization of FAR1 is preserved but that the plasmalogen-dependent regulation of FAR1 protein levels is abolished, resulting in uncontrolled ether lipid synthesis and considerable changes in the cellular lipidome.
Previously, biallelic loss-of-function variants in FAR1 have been reported in a limited number of patients with FAR1 deficiency. Of the five previously reported patients, all had (profound) intellectual disability, severe growth retardation/failure to thrive, microcephaly, spasticity, and facial dysmorphism. Four of five patients had congenital cataracts and two had cerebral white matter abnormalities.8,9 Patients with FAR1 heterozygous de novo variants share many clinical symptoms with FAR1 deficiency, including spastic features, seizures, and bilateral cataracts, but have normal brain magnetic resonance images (MRIs), normal growth, no microcephaly, and no dysmorphic features. Nevertheless, the biochemical phenotypes are the complete opposite of each other; the FAR1-deficient patients have a profound deficiency of plasmalogens whereas the patients with the heterozygous de novo variants have elevated levels of ether lipids, including plasmalogens. Diseases with both an autosomal dominant and autosomal recessive inheritance pattern in the same gene (e.g., VLCFA elongase 4 deficiency, ELOVL416) and diseases caused by either loss- or gain-of-function variants in the same gene have been described before (in, e.g., SCN9A17 and SLC40A118); however, all have either different clinical or similar biochemical phenotypes. The combination of an overlapping clinical phenotype caused by either biallelic variants or de novo heterozygous variants with a completely opposite biochemical phenotype has, to our knowledge, hitherto not been observed before.
FAR1 p.Arg480His/Cys variants disturb the plasmalogen-dependent modulation of FAR1
When plasmalogen levels are high, FAR1 activity should be downregulated by increased degradation of FAR1 protein as part of an elegant and effective feedback mechanism in the ether lipid biosynthetic pathway.19 We showed that the FAR1 p.Arg480His/Cys variants abolish the plasmalogen-dependent modulation of FAR1 protein levels, which eliminates this negative feedback mechanism and results in considerably elevated ether lipid synthesis. Arginine 480 is located in the transmembrane domain of FAR1 (formed by amino acids 466–483), close to the border of this domain facing the peroxisomal matrix. Our study identifies this arginine as an essential amino acid in this feedback regulatory mechanism, but how and why the change to another amino acid prevents the degradation of FAR1 in response to increased plasmalogen levels is unclear. Nevertheless, our results are in line with data from Honsho et al.7 who showed that the C-terminal domain of FAR1, especially the 17 amino acids flanking the transmembrane domain (491–507), is essential for the plasmalogen-dependent modulation of the stability of FAR1.7
Disruption of both the ether/nonether lipid balance as well as the fatty acid distribution
Our lipidomics analysis revealed that in addition to changes in plasmalogen levels there were also profound changes in the composition of cellular lipids. We noted elevated levels of ether lipids (PC[O], PE[O], DG[O], and TG[O]) with a concomitant reduction of the nonether lipid PC and (L)PE. One possible mechanism for the observed shift is that high levels of DG[O] compete with DG for the limited amounts of activated headgroups CDP-ethanolamine and CDP-choline to synthesize ether and nonether analogs of PC and PE, shifting the balance toward ether phospholipids at the expense of the nonether equivalents. Interestingly, this ether/nonether balance is precisely reversed in different RCDP subtypes where nonether lipids compensate for the absence of their ether counterparts.20 The accumulation of DG[O] likely also causes the enhanced synthesis of the minor lipid TG[O]. In addition to this shift in the major lipid classes there also was an effect on the fatty acid composition. PUFA-containing species appeared to be redistributed away from PC, PS, and CL toward PC[O] and TG. In different types of RCDP the opposite is true; nonether counterparts contain higher PUFA levels,20 which parallels the changes in PC/PC[O] and PE/PE[O] mentioned above. Ether (phospho)lipids have different physicochemical properties when compared with their nonether analogs and are handled differently through the biosynthetic and remodeling systems. Their fatty acid content is also different in that ether phospholipids are enriched in PUFAs such as docosahexaenoic acid (C22:6n-3) and arachidonic acid (C20:4n-6), which is achieved by synthesis and remodeling.1 The changes in fatty acid composition could be a way for the cell to balance membrane fluidity caused by the shift in the ether/nonether lipid balance.
Ether lipids and fatty alcohols: a delicate balance?
The two most consistent clinical symptoms in this cohort were spastic paraparesis and bilateral cataracts. Remarkably, cataracts are also observed in FAR1 deficiency, other forms of RCDP, and peroxisome biogenesis disorders that are characterized by a primary or secondary deficiency of ether lipid synthesis. In a mouse model for RCDP type 1 (Pex7 mouse model) with profound plasmalogen deficiency, restoration of plasmalogen levels by alkylglycerol supplementation prevented cataract formation establishing that the shortage of ether lipids causes the cataracts.21 On the other side of the spectrum, three disorders have recently been reported that biochemically show elevated levels of ether lipids: EPT1 deficiency (SELENOI),22,23 PCYT2 deficiency (PCYT2),14,24 and Sjögren–Larsson syndrome (SLS, ALDH3A2).25,26 Like the FAR1 p.Arg480 variant patients, spastic paraparesis and some form of ocular pathology (e.g. cataracts, optic atrophy, macular dystrophy) are frequently observed in these disorders. It is intriguing that both the shortage of ether lipids and the excessive accumulation of this class of lipids results in similar pathology. Besides ether lipids, fatty alcohols possibly are involved in the pathological mechanism. Fatty alcohols have been shown to accumulate in SLS25,26 and RCDP types 1–327 and may accumulate in PCYT2 and EPT1 deficiency given the disturbed synthesis of ethanolamine ether lipids,14 but would obviously not accumulate in FAR1 deficiency where they cannot be synthesized. Despite the similarities and differences in symptoms and biochemical abnormalities, the exact pathophysiological mechanism remains enigmatic. It is obvious, however, that a disturbance of the fatty alcohol cycle and the linked process of ether lipid synthesis is involved in the neurological and ocular pathology of these disorders. Our findings underline that both fatty alcohol and ether lipid levels need to be tightly regulated, because an imbalance leads to disease.
Conclusion
We describe an autosomal dominant disorder for the known peroxisomal disease gene FAR1 with an overlapping phenotype but a different underlying disease mechanism than that of recessive FAR1 deficiency. Our findings provide fundamental clues about the regulation of ether lipid synthesis by FAR1 protein levels and show that for patients with spastic paraparesis and cataracts, FAR1 should be considered as a candidate gene and added to gene panels for hereditary spastic paraplegia, cerebral palsy, and juvenile cataracts. Finally, the measurement of plasmalogens in erythrocytes should be considered as a functional readout of ether lipid synthesis as both abnormally high or low levels could suggest FAR1-related pathology.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and/or its supplementary material or can be made available upon reasonable request.
Change history
19 October 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41436-021-01189-8
References
Braverman, N. E. & Moser, A. B. Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta 1822, 1442–1452 (2012).
Dean, J. M. & Lodhi, I. J. Structural and functional roles of ether lipids. Protein Cell 9, 196–206 (2018).
Brites, P., Waterham, H. R. & Wanders, R. J. A. Functions and biosynthesis of plasmalogens in health and disease. Biochim. Biophys. Acta 1636, 219–231 (2004).
Wanders, R. J. A. Metabolic functions of peroxisomes in health and disease. Biochimie 98, 36–44 (2014).
Cheng, J. B. & Russell, D. W. Mammalian wax biosynthesis. I. Identification of two fatty acyl-Coenzyme A reductases with different substrate specificities and tissue distributions. J. Biol. Chem. 279, 37789–37797 (2004).
Honsho, M., Asaoku, S. & Fujiki, Y. Posttranslational regulation of fatty acyl-CoA reductase 1, Far1, controls ether glycerophospholipid synthesis. J. Biol. Chem. 285, 8537–8542 (2010).
Honsho, M., Asaoku, S., Fukumoto, K. & Fujiki, Y. Topogenesis and homeostasis of fatty acyl-CoA reductase 1. J. Biol. Chem. 288, 34588–34598 (2013).
Buchert, R. et al. A peroxisomal disorder of severe intellectual disability, epilepsy, and cataracts due to fatty acyl-CoA reductase 1 deficiency. Am. J. Hum. Genet. 95, 602–610 (2014).
Alshenaifi, J. et al. The many faces of peroxisomal disorders: lessons from a large Arab cohort. Clin. Genet. 95, 310–319 (2019).
Duran, M. & Wanders, R. J. A. in Laboratory Guide to the Methods in Biochemical Genetics (eds Blau, N., Duran, M. & Gibson, K. M.) 207–220 (Springer, Berlin Heidelberg, 2008).
Ferdinandusse, S. et al. A novel case of ACOX2 deficiency leads to recognition of a third human peroxisomal acyl-CoA oxidase. Biochim. Biophys. Acta 1864, 952–958 (2018).
Ferdinandusse, S. et al. ACBD5 deficiency causes a defect in peroxisomal very long-chain fatty acid metabolism. J. Med. Genet. 54, 330–337 (2017).
Exner, T. et al. An alternative membrane topology permits lipid droplet localization of peroxisomal fatty acyl-CoA reductase 1. J. Cell Sci. 132, jcs223016 (2019).
Vaz, F. M. et al. Mutations in PCYT2 disrupt etherlipid biosynthesis and cause a complex hereditary spastic paraplegia. Brain 142, 3382–3397 (2019).
Sobreira, N., Schiettecatte, F., Valle, D. & Hamosh, A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 36, 928–930 (2015).
Deák, F., Anderson, R. E., Fessler, J. L. & Sherry, D. M. Novel cellular functions of very long chain-fatty acids: insight from ELOVL4 mutations. Front. Cell. Neurosci. 13, 428 (2019).
Drenth, J. P. H. & Waxman, S. G. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J. Clin. Invest. 117, 3603–3609 (2007).
Callebaut, I. et al. Comprehensive functional annotation of 18 missense mutations found in suspected hemochromatosis type 4 patients. Hum. Mol. Genet. 23, 4479–4490 (2014).
Honsho, M., Abe, Y. & Fujiki, Y. Plasmalogen biosynthesis is spatiotemporally regulated by sensing plasmalogens in the inner leaflet of plasma membranes. Sci. Rep. 7, 43936 (2017).
Dorninger, F. et al. Homeostasis of phospholipids - the level of phosphatidylethanolamine tightly adapts to changes in ethanolamine plasmalogens. Biochim. Biophys. Acta 1851, 117–128 (2015).
Brites, P. et al. Alkyl-glycerol rescues plasmalogen levels and pathology of ether-phospholipid deficient mice. PLoS ONE 6, e28539 (2011).
Ahmed, M. Y. et al. A mutation of EPT1 (SELENOI) underlies a new disorder of Kennedy pathway phospholipid biosynthesis. Brain 140, 547–554 (2017).
Horibata, Y. et al. EPT1 (selenoprotein I) is critical for the neural development and maintenance of plasmalogen in humans. J. Lipid Res. 59, 1015–1026 (2018).
Vélez-Santamaría, V. et al. Expanding the clinical and genetic spectrum of PCYT2-related disorders. Brain 143, e76 (2020).
S’aulis, D., Khoury, E. A., Zabel, M. & Rizzo, W. B. 1-O-Alkylglycerol accumulation reveals abnormal ether glycerolipid metabolism in Sjögren-Larsson syndrome. Mol. Genet. Metab. https://doi.org/10.1016/j.ymgme.2020.08.002 (2020).
Staps, P. et al. Disturbed brain ether lipid metabolism and histology in Sjögren-Larsson syndrome. J. Inherit. Metab. Dis. https://doi.org/10.1002/jimd.12275 (2020).
Rizzo, W. B. et al. Fatty alcohol accumulation in the autosomal recessive form of rhizomelic chondrodysplasia punctata. Biochem. Med. Metab. Biol. 50, 93–102 (1993).
Acknowledgements
The authors thank Elise van der Sluijs, Henny Rusch, Henk van Lenthe, and Serhii Chornyi for technical assistance and Clara van Karnebeek and Carlos Ferreira for helpful discussions. The Core Facility Metabolomics Amsterdam UMC is acknowledged for the lipidomics studies. This work was supported in part by the Common Fund, Office of the Director, National Institutes of Health (NIH) and the Intramural Research Program of the National Human Genome Research Institute, NIH (Bethesda, MD, USA). Portions of this work were supported by 1R01 NS106298.
Author information
Authors and Affiliations
Consortia
Contributions
Conceptualization: S.F., K.M., L.I.J., F.V.; Formal Analysis: K.M., M.P., E.W., F.M., M.S., A.B., Q.W., S.B., A.K.; Investigation: M.T., L.B., R.L., S.S., C.N., J.D., C.T., E.U., S.P., A.D., K.C., N.M., J.B., E.B., C.T., W.G., E.M., L.W., U.D.N., P.Z., A.Z., M.B., R.S., S.E., D.G., M.K., D.C.; Methodology: H.B., P.M., J.R., A.L.; Project administration: S.F., K.M., F.V.; Resources: M.T., L.B., R.L., S.S., C.N., J.D., C.T., E.U., S.P., A.D., K.C., N.M., J.B., E.B., C.T., W.G., E.M., L.W., U.D.N., P.Z., A.Z., M.B., R.S., S.E., D.G., M.K., D.C.; Supervision: S.F., K.M., F.V.; Visualization: H.B., P.M., F.V.; Writing – original draft: S.F., K.M., R.W., H.W., D.C., F.V.; Writing – review & editing: S.F., K.M., H.B., L.IJ, P.M., J.R., A.L., M.P., E.W., F.M., M.S., A.B., M.T., L.B., R.L., S.S., C.N., J.D., C.T., E.U., S.P., A.D., K.C., N.M., J.B., E.B., C.T., W.G., E.M., L.W., U.D.N., Q.W., P.Z., A.Z., M.B., R.S., S.E., D.G., S.B., M.K., A.K., R.W., H.W., D.C., F.V.
Corresponding authors
Ethics declarations
Competing interest
K.M., A.B., F.M., M.J.G.S. are employees of GeneDx, Inc. A.V.D. is an advisory board member for ProQr. The other authors declare no competing interests.
Ethics declaration
Informed consent was obtained from the legal guardians of the patients described in this study; for specifics, see Supplementary information 1. The fibroblasts of healthy controls subjects were collected for research purposes after informed consent and approval by the medical ethics committee of the Academic Medical Center in Amsterdam.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Unfortunately Seth Perlman erroneously credited as having a PhD in the authors list. Seth Perlman have only an MD.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ferdinandusse, S., McWalter, K., te Brinke, H. et al. An autosomal dominant neurological disorder caused by de novo variants in FAR1 resulting in uncontrolled synthesis of ether lipids. Genet Med 23, 740–750 (2021). https://doi.org/10.1038/s41436-020-01027-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-01027-3
This article is cited by
-
Complex Hereditary Spastic Paraparesis Caused by de novo p.Arg480Ser in FAR1
Indian Journal of Pediatrics (2024)
-
A conserved complex lipid signature marks human muscle aging and responds to short-term exercise
Nature Aging (2024)
-
Post-Bariatric Hypoglycemia: an Impaired Metabolic Response to a Meal
Obesity Surgery (2024)
-
Mendelian inheritance revisited: dominance and recessiveness in medical genetics
Nature Reviews Genetics (2023)
-
Metabolomic analysis of maternal mid-gestation plasma and cord blood in autism spectrum disorders
Molecular Psychiatry (2023)
