
Abstract
Purpose
To determine the diagnostic yield and clinical impact of exome sequencing (ES) in patients with suspected monogenic kidney disease.
Methods
We performed clinically accredited singleton ES in a prospectively ascertained cohort of 204 patients assessed in multidisciplinary renal genetics clinics at four tertiary hospitals in Melbourne, Australia.
Results
ES identified a molecular diagnosis in 80 (39%) patients, encompassing 35 distinct genetic disorders. Younger age at presentation was independently associated with an ES diagnosis (p < 0.001). Of those diagnosed, 31/80 (39%) had a change in their clinical diagnosis. ES diagnosis was considered to have contributed to management in 47/80 (59%), including negating the need for diagnostic renal biopsy in 10/80 (13%), changing surveillance in 35/80 (44%), and changing the treatment plan in 16/80 (20%). In cases with no change to management in the proband, the ES result had implications for the management of family members in 26/33 (79%). Cascade testing was subsequently offered to 40/80 families (50%).
Conclusion
In this pragmatic pediatric and adult cohort with suspected monogenic kidney disease, ES had high diagnostic and clinical utility. Our findings, including predictors of positive diagnosis, can be used to guide clinical practice and health service design.
Similar content being viewed by others

INTRODUCTION
Chronic kidney disease (CKD) affects more than 10% of the adult population and causes substantial morbidity and mortality, primarily related to the increased risk of cardiovascular disease.1 Genetic kidney disease (GKD) is increasingly recognized as an important cause of CKD and can be difficult to accurately diagnose using traditional diagnostic strategies.2 Genomic studies have provided estimates of the prevalence of GKD, demonstrating a diagnostic yield of almost 10% in unselected adults3 and 20–30% in children with nephropathy.4,5 The clinical application of genomic technologies has the potential to transform diagnostic pathways by providing a timely and accurate genetic diagnosis.6 In patients with GKD, this has several potential benefits, including providing prognostic information, informing targeted surveillance and therapies,7,8,9 preventing inappropriate treatments,10 informing reproductive decisions, and reducing the use of invasive diagnostic investigations such as renal biopsies.
Genomic testing is becoming established as a powerful diagnostic tool in pediatric rare disease, and evidence that genomic testing informs clinical decisions is beginning to emerge in this context.11,12 To inform implementation across other specialties, such as nephrology, it is important to specifically evaluate the diagnostic and clinical utility of genomic tests in relevant patient groups and real-world contexts. The largest study of genomic testing in renal patients to date reported a diagnostic yield of just under 10% in an unselected cohort.3 The study was research-based, with only broad categories and diagnostic codes available in terms of phenotypic data for a substantial proportion of patients. Results were not returned to patients, and hence the clinical value of genomic testing, both to the patient and their family, was not evaluated. A small number of studies have provided retrospective or pilot single-center evaluations of clinical genomic tests in renal patients13,14 or evaluated research-based genomic tests in particular subgroups of kidney disease such as transplant recipients or steroid-resistant nephrotic syndrome.4,15 This preliminary evidence supports the value of exome sequencing (ES) in both adult and pediatric patients with renal disease, paving the way for wider implementation.
Such broader implementation poses significant challenges, particularly with regard to developing evidence-based guidelines for patient triage and testing, as well as the development of service delivery models that enable timely access to genetic counseling, specialists, and testing services. Renal genetics clinics (RGC) are one model of addressing these challenges. RGCs apply a multidisciplinary team (MDT) approach through collaboration between adult and pediatric nephrologists, clinical geneticists, genetic counselors, and laboratory scientists. Preliminary evidence has shown that dedicated RGCs may improve diagnostic outcomes and enhance care of patients.14,16,17
We sought to determine the diagnostic yield and clinical impact on patients and their families of clinically accredited ES in a prospective cohort of adults and children with suspected genetic kidney disease in the context of a multidisciplinary RGC.
MATERIALS AND METHODS
Ethics statement
This study was approved by the Human Research Ethics Committee at Melbourne Health (2016.224) and site-specific research governance approval was obtained from all participating hospitals. All patients or their guardians provided written informed consent for clinical ES and participation in the study.
Study design
This prospective multicenter cohort study recruited patients referred to one of four RGC teams based at tertiary hospitals in Melbourne, Australia between July 2017 and September 2018, with data collected to October 2019. All reporting was performed in accordance with the Strengthening the Reporting of Observational Studies in Epidemiology guidelines.18
Following referral by their treating nephrologist, patients were discussed for study inclusion by the RGC team. Patients were recruited if their clinical presentation was consistent with a monogenic cause (e.g., glomerular, tubulointerstitial, or cystic renal disease) and prioritized if they met one of the following criteria: family history of renal disease, syndromic features, or childhood onset of disease. Patients who did not meet these criteria were considered at the discretion of the MDT. Patients with phenotypes where the likelihood of a monogenic cause is low, for example, congenital anomalies of the kidney and urinary tract (CAKUT) were only included if they had extrarenal features. Patients with a pre-existing molecularly confirmed genetic diagnosis or a phenotype and family history suggestive of typical autosomal dominant polycystic kidney disease (ADPKD) were excluded.
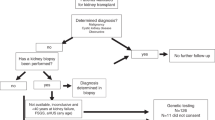
Patients were reviewed in a multidisciplinary RGC setting by a nephrologist, clinical geneticist, and genetic counselor on at least two occasions (before and after testing). Chromosomal microarray (CMA) was recommended in all patients prior to ES to identify copy-number variants (CNVs). The workflow from initial patient review to return of results is summarized in Fig. 1.

Referral received by nephrologist involved in multidisciplinary RGC. Triage of referral by multidisciplinary team (MDT) consisting of nephrologist/s, clinical geneticist, and genetic counselor. Patient attends for clinic appointment for clinical review, pretest genetic counseling, and consent. Sample collected at local hospital. Genomic data isolated from blood and saliva samples. Clinically accredited genomic sequencing performed. Initial computational bioinformatics analysis. Variant prioritization meeting to prioritize variants for assessment, using a tiered approach, attended by a senior medical genomic scientist, clinical geneticists, lead nephrologist, referring nephrologist from RGC, and genetic counselors. Variant curation as per American College of Medical Genetics and Genomics (ACMG) guidelines. Review of variant and phenotypic data by MDT of laboratory scientists, clinical geneticists, nephrologists from the RGC and genetic counselors to reach consensus prior to reporting and ensure that genotype is concordant with phenotype. Patient attends clinic appointment for return of results with post-test genetic counseling, with segregation encouraged (where appropriate), suitable patients flagged for recruitment into research. *See “Materials and Methods” for details, including outlined of tiered approach to testing.
Data collection
Detailed clinical and demographic data were collected and entered into a secure REDCap19,20 electronic data capture tool hosted at the Murdoch Children’s Research Institute. Comprehensive phenotypic information was entered prospectively by the nephrologist, including the presence of extrarenal features, kidney function, blood pressure, hematuria, proteinuria, biochemical/hematological abnormalities, imaging, renal histopathology, and results of prior genetic investigations if already performed. A detailed family history was recorded to identify the number of at-risk relatives.
The clinically suspected diagnosis and differential diagnoses were collected at two different time points prior to release of ES results: (1) the suspected diagnoses by the referring physician and (2) the suspected diagnoses following review by the RGC team. Additional details, including diagnostic tests that may have been performed in the absence of ES, were also collected. Diagnostic utility for each patient was recorded: we classified patients as having a “confirmation of diagnosis” when the referral diagnosis was concordant, and “clarification of diagnosis” when ES eliminated diagnostic or inheritance uncertainty (in cases where the presumed inheritance pattern was incorrect or unknown, or where there were up to two possible differential diagnoses listed). Recommendations and changes to clinical management were recorded by nephrologists at return of ES results and at three months following results.
Exome sequencing and variant analysis
Genomic DNA was isolated from blood and saliva samples. ES was performed in a clinically accredited laboratory (Victorian Clinical Genetics Services, Melbourne). Coding regions were enriched using the CREv2 exome capture kit (Agilent). Sequencing was performed on HiSeq 4000 (Illumina) to an average target depth of 100×. Data were analyzed using an in-house validated version of Cpipe,21 followed by variant filtering and analysis using an in-house adapted version of the Leiden Open Variation Database (LOVD).22,23 A variant prioritization meeting was attended by a laboratory scientist, clinical geneticists, genetic counselors, and nephrologists. Data were analyzed only for variants in conditions relating to patient phenotype following a tiered approach. Initially, variants in genes associated with the patient’s specific disease category (e.g., glomerular disease) were evaluated. If no variants were identified, analysis was expanded to a broader group of 336 known kidney disease genes.24 These are listed in Supplementary table S1. Where clinically relevant (in patients with extrarenal manifestations suggestive of a syndromic diagnosis), a virtual panel of ~4000 genes known to cause Mendelian disorders (Mendeliome) was used.25 Standard turnaround time for ES was six months. Nine patients had had rapid ES processing based on clinical need, with results available within 21 days. Variants were classified according to the current American College of Medical Genetics and Genomics (ACMG) guidelines for clinical sequencing interpretation.26,27 Variant and phenotypic data were reviewed at a genomics MDT to reach consensus on variant and case-level interpretation prior to reporting. Segregation was pursued in cases where the additional information would alter classification of the variant.
Outcomes
The primary outcome was the ES diagnostic yield. Molecular diagnosis was compared with suspected clinical diagnosis at referral and following RGC assessment. The diagnostic yield was compared between clinical diagnostic subgroups. The secondary outcome was diagnostic and clinical utility of ES. Only ES diagnoses that explained the renal phenotype contributed to the diagnostic yield.
Statistical analysis
Baseline characteristics were expressed as frequencies (n, %) and medians (range), as dictated by the data type. Chi-squared and Fisher’s exact tests (FET) were performed to compare receiving a positive ES diagnosis and various clinical characteristics. Multivariable logistic regression was performed to identify baseline clinical characteristics associated with a diagnostic result. Candidate explanatory variables were identified from a review of the literature. The final multivariable model included all biologically plausible variables that were robustly collected. Prespecified first-order interaction terms between covariates were examined. Linearity assumptions were validated by dividing continuous data into quartiles and fitting as categorical variables. Data were analyzed using Stata 15.1 (College Station, TX). Two-sided p values < 0.05 were considered statistically significant.
RESULTS
A total of 225 probands met our inclusion criteria; 21 were excluded as they declined to participate, withdrew consent, did not provide a sample for analysis, or obtained a diagnosis via CMA and did not proceed to ES (Fig. 2). ES was performed in 204 patients (81 pediatric and 123 adults). Most patients (147/204) had CMA performed prior to ES, which was uninformative. Few patients had other forms of genetic testing prior to the study (five patients had single-gene testing and four patients had panel testing, all of which were also uninformative). One patient (P132) had a diagnostic CMA that was performed concurrently to ES (Supplementary table S5). The median age at the time of recruitment was 28.5 years (range 1 month–72 years). The median age at first presentation to nephrology was 16 years (range 0 months–64 years). A total of 138 (68%) had self-declared Caucasian ethnicity, consanguinity was reported in 11 families (5%), and 103 patients (50%) had a family history of renal disease. Clinical characteristics of this cohort are provided in Table 1. At least one of the three major inclusion criteria (family history of renal disease, presentation of renal disease in childhood, or extrarenal manifestations) was present in 182/204 (89%). ES analysis was limited to genes known to cause GKD (Supplementary table S1) for 182 patients and Mendeliome analysis was performed in 22 patients. The median time from ES request to result report was 6 months (range 10 days to 15.8 months) and the median time from referral to review in the RGC was 2.4 months (range: 0 months to 16.9 months).

CMA chromosomal microarray, ES exome sequencing.
Genomic findings and diagnostic yield
A molecular diagnosis was obtained in 80 of the 204 (39%) patients (Table 2 and Supplementary table S2). All variants have been deposited into ClinVar28 (accession numbers SCV001427100–SCV001427205). Of the 35 monogenic disorders identified, pathogenic and likely pathogenic variants in seven genes accounted for 50 of the 81 genetic diagnoses (one patient had two genetic diagnoses). These included COL4A3 (n = 6), COL4A4 (n = 9), COL4A5 (n = 11) associated with Alport syndrome–related nephropathy; HNF1B (n = 5) associated with renal cysts and diabetes syndrome (RCAD); PKD1 (n = 9) and PKD2 (n = 1) associated with ADPKD; and PKHD1 (n = 9) associated with autosomal recessive polycystic kidney disease (ARPKD) (Supplementary figure 1). In five patients with two heterozygous variants found in autosomal recessive genes, it was not possible to confirm the variants were in trans within the study timeframe (supplementary table S2). The variants were assumed to be in trans when there was a clear genotype–phenotype correlation and patients were counseled as to the caveats of diagnosis.
In addition, we identified potentially clinically relevant variants of uncertain significance (VUS) that did not meet criteria to be reported as likely pathogenic or pathogenic in 52 patients (25%) (Supplementary table S3). All VUS were not considered to be clinically actionable and patients were counseled accordingly. In these cases, a letter was sent to the patient’s treating nephrologist, encouraging them to refer to the RGC in 2–3 years, for consideration of genomic data reanalysis.
Clinical predictors of positive ES diagnosis for kidney disease
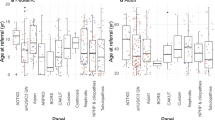
The diagnostic yield of ES was higher in the pediatric group, with 38/81 (47%) children compared with 42/123 (34%) adults receiving a molecular diagnosis (χ2 = 3.34, p = 0.07). Patients with disease onset in childhood had a higher diagnostic yield compared with those who presented in adulthood (56/110 = 51% versus 24/94 = 26%, (χ2 = 13.69, p = 0 < 0.001) regardless of age at recruitment.
The diagnostic yield of ES differed by diagnostic subgroup (p < 0.001, FET), being highest in those with tubulopathy (11/18 [61%)]), Alport syndrome (24/43 [63%]), and cystic kidney disease (31/65 [48%]). A molecular diagnosis was reported in 3/14 (21%) with syndromic CAKUT, 7/39 (18%) with nephrotic syndrome, and 4/14 (29%) with nephropathy attributed to other causes. No patients with a clinical diagnosis of a complement disorder (n = 6) or nephropathy of unknown origin (n = 5) received a genomic diagnosis (Table 3). In 70/80 patients receiving an ES diagnosis (88%), the diagnostic variants were identified within the virtual gene panel assigned based on the referral clinical diagnosis group (e.g., glomerular renal disease), and the remainder of the diagnoses (10/80, 12%) were obtained from expanded analysis.
On multivariable analysis age at first presentation of renal disease and family history were independent clinical predictors of ES diagnosis (Supplemental Fig. 2 and Supplementary table S4). There was no association between female sex, presence of extrarenal manifestations, or parental consanguinity with a positive diagnosis. Compared with those with onset of renal disease after 30 years of age, those with renal disease in infancy (<1 years; odds ratio [OR] 9.05, 95% confidence interval [CI] 2.90–28.26) or childhood (age 1 to 17 years; OR 2.42, 95% CI 1.21–6.24) were more likely to have a molecular diagnosis on ES (p < 0.001). Patients with a positive family history of renal disease in at least one first or second degree relative were more likely to receive ES diagnosis (OR 2.51, 95% CI 1.21–4.84, p = 0.01).
Diagnostic and clinical utility of ES
When comparing the suspected clinical diagnosis at referral to RGC in the 80 patients with ES diagnoses, 27 (34%) had their clinical diagnosis confirmed, 22 (28%) had clarification of their diagnosis, and 31 (39%) had complete reclassification of their original diagnosis (Fig. 2, Supplementary table S2). ES diagnosis had management implications in 47/80 patients [59%]). These included negating the need for diagnostic renal biopsy in 10/80 (13%), changing surveillance in 35/80 (44%), and changing treatment plan in 16/80 (20%). Other specific changes to management were made in 7/80 (9%), which included facilitating an earlier transplant, preventing invasive investigations, and informing decision making in pregnancy. Reproductive counseling was not included as a clinical implication unless it resulted in a specific outcome. Even in cases with no changes to management in the proband, the ES result had implications for the management of family members in 26/33 (79%) cases. Cascade testing was offered to 40/80 (50%) families and changes in management were initiated in family members of 23/80 patients with a diagnosis. Table 2 summarizes the clinical utility of all patients with an ES diagnosis. Details of all diagnostic genetic variants and their clinical utility are outlined in Supplementary table S2. In addition, a further five patients in the study achieved a molecular diagnosis but were not included in the total diagnostic yield (Supplementary table S5).
ES proved valuable even in cases where a suspected diagnosis was confirmed or clarified. For example, in 16 patients with suspected hereditary nephritis or Alport syndrome, ES informed surveillance in all patients (e.g., audiology and ophthalmology screening or changed frequency of nephrology review), allowed identification of at-risk relatives who were subsequently offered genetic testing in 11 families (Table 2), and obviated the need for diagnostic renal biopsy in 7 patients. In another patient, P137, ES confirmed the suspected diagnosis of autosomal dominant hypocalciuric hypercalcemia. This patient had previously undergone parathyroidectomy, which would not have occurred if this diagnosis was known. The patient was provided reassurance regarding ongoing hypercalcemia postsurgery and her at-risk child was subsequently referred for biochemical screening.
In 31/80 patients (39%), ES reclassified the clinical diagnosis. In patient P130, genomic testing revised a longstanding clinical diagnosis of ADPKD to ARPKD. This diagnosis provided valuable information with regard to family planning, with retraction of the previously given 50% recurrence risk. The patient conceived naturally, and a year later, had an unaffected child. In P020, ES diagnosed autosomal recessive Alport syndrome in a child who presented with steroid-resistant nephrotic syndrome and microscopic hematuria. Renal biopsy suggested an inherited glomerular basement membrane abnormality. Following ES result, a recommendation was made to cease existing cyclosporin treatment, with renin–angiotensin inhibition, audiology, and ophthalmology surveillance to be continued; his siblings were referred for cascade testing.
Rapid genomic testing
Nine patients underwent rapid ES, with median time to report 28 days (range 10–49 days). The characteristics and results of this group are outlined in Supplementary table S6. In five cases, rapid ES resulted in direct changes to clinical management. For example, a seven-year-old boy who was initially thought to have Dent disease developed heavy proteinuria and a renal biopsy was planned for suspected Alport syndrome. Rapid ES confirmed X-linked recessive (XLR) Dent disease, negating the need for renal biopsy and audiology and ophthalmology surveillance. Cascade genetic testing was subsequently offered for his two brothers. In another patient with features of an undiagnosed ciliopathy, rapid ES diagnosed autosomal recessive short-rib thoracic dysplasia 10 with or without polydactyly (OMIM 615630). This not only provided important prognostic information including an explanation for the multisystem nature of the disease, but it also clarified that her father was at low risk of the same condition, enabling live kidney donation.
DISCUSSION
In this large, prospective multicenter study, we comprehensively evaluated the diagnostic and clinical utility of ES in adults and children with suspected genetic CKD. Our cohort was ethnically diverse, and diagnostic variants were detected within all major clinical diagnostic subcategories (such as Alport, nephrotic syndrome, tubulopathies, and cystic kidney disease). By using a multidisciplinary approach in concert with clinical ES, we detected a monogenic cause of kidney disease in 39% of patients. Our diagnostic yield in adults (34%) is significantly higher than the recently published cohort by Groopman et al.,3 which likely reflects the differences in patient selection, as patients in this study were assessed as having possible GKD following MDT assessment. Our yield is comparable with other published cohorts with suspected GKD,13,29,30 apart from the retrospective study by Thomas et al.,14 which found a diagnosis in 26/43 (60%) patients who underwent genetic testing. The lower diagnostic yield in our cohort is likely due to several reasons. First, as our study focus was on ES, we did not include patients who presented to RGC for cascade testing in the context of an established genetic diagnosis in the family; these patients were more suitable for Sanger sequencing. Second, the study by Thomas et al. included a large proportion of patients with clinically suspected ADPKD. ADPKD is the leading cause of Mendelian kidney disease and can be confidently diagnosed clinically.31 We did not intend to include those patients with an already established ADPKD diagnosis and hence patients with a phenotype and family history highly suggestive of typical ADPKD were excluded from our cohort. Finally, our cohort was much larger and more diverse, and did not include multiple patients from the same pedigree.
This is the only prospective study to date that provides detailed evaluation of both diagnostic and clinical utility of ES in kidney disease patients. Apart from the small retrospective study by Thomas et al., no other studies have evaluated the actual clinical implications of ES for patient and family management. While some studies have attempted to infer the potential clinical implications of a genomic diagnosis on patients,2,13 they did not collect detailed information to measure the actual implications for each individual patient. ES altered the diagnosis in a substantial proportion of individuals in our cohort (39%), compared with recently published studies.13,16 Even in cases where ES confirmed a clinical diagnosis, this proved valuable by providing a definitive diagnosis and information on inheritance pattern, thereby preventing further investigations and enabling appropriate screening and genetic counseling of family members. Patients with a revised diagnosis following ES experienced even more significant changes in clinical management, including avoidance of immunosuppression, avoidance of renal biopsy, appropriate medical surveillance, cascade testing and screening among family members, and informing transplant and reproductive decisions.
One of the most important benefits of ES in the renal setting may be avoiding unnecessary and invasive diagnostic investigations such as renal biopsy, which carry considerable risk and cost, particularly in the pediatric population.32,33 In our study, a planned renal biopsy was no longer required in ten patients who received a molecular genetic diagnosis. Genomic testing has already been shown to be cost-effective in other specialties, such as pediatric acute care.34 During this study period, the cost of ES was approximately AU$2300, or US$1600 (including patient review, sequencing, and analysis of up to 100 genes). The cost of a biopsy varies, but an uncomplicated renal biopsy is approximately AU$1600 (US$1100) per adult and AU$5300 (US$3700) per child at our institution. Therefore, a timely genomic diagnosis has the potential to be cost-effective, particularly in those patients where biopsies are no longer required. In some cases, the standard turnaround time (TAT) for genomic results achieved during this study of six months is likely to be too long. For this reason, we performed rapid sequencing (with a result available within a week if required) in those patients where it was predicted to have immediate clinical implications. This TAT had already improved to 3 months by the end of the study, mainly due to improved experience among clinicians, resulting in increased efficiency at variant prioritization meetings and MDT meetings, and is likely to improve further as genomic testing practices become more streamlined.
Our study provides data to assist with prioritizing patients for genomic testing, particularly in resource-limited health systems. In our cohort, age at presentation of renal disease and positive family history were independent clinical predictors of ES diagnosis. Extrarenal features, which have been recognized as risk factors for genetic disease in previous studies,29 were not significantly associated with ES diagnosis in this cohort. Most patients with extrarenal features (40/53) presented in childhood, hence it is possible that in this cohort childhood onset was a proxy for extrarenal manifestations. There was no association with an ES diagnosis and parental consanguinity, although it is possible that our study may be underpowered to detect a difference, due to the small number of patients (n = 11) with this history. Patients with cystic, Alport, and tubulopathy phenotypes had the highest diagnostic yield, suggesting that ES is a particularly useful diagnostic test in these groups. When interpreting the diagnostic yield, it is important to note that all our patients were referred for clinical rather than for research-based testing. Therefore, our results are pragmatic and are more likely to be replicated in clinical practice compared with studies that conducted research-based testing. We also recognize that the diagnostic yield may improve in future with reanalysis of ES data,35 and this has recently been funded by our health-care system. Compared with the few other studies that have applied genomic testing to similar cohorts,2,13,30 our study collected detailed phenotypic information, including a specific a priori clinical diagnosis and all prior investigations. This allowed for a more accurate determination of phenotype–genotype correlation. Furthermore, to minimize bias, the physicians who recorded implications for clinical care were those directly involved with each patient, rather than the main study investigators.
Many barriers to effective implementation of genomics in nephrology and in other specialties have been described,36 including physician knowledge gaps surrounding genetics.37 Knowledge gaps among nephrologists surrounding assessment of patients with GKD, testing processes, patient consent, and counseling were also an important implementation challenge in our study. None of the nephrologists involved in the genetics clinics in this study had formal genetics training, although three had additional research and clinical experience in this area. This challenge was foreseen prior to study commencement, and the multidisciplinary RGC model, which allowed support from a clinical geneticist and genetic counselor in reviewing all patients, was used to address this.38 Our study has limitations. We did not consider the possible contribution of digenic inheritance or susceptibility alleles to the diagnostic yield in this cohort. ES has limited utility in detecting structural, mitochondrial, and noncoding variants.39 Clinicians were encouraged to perform CMA prior to ES in all patients. As microarray is performed routinely as part of the initial diagnostic work-up of patients with suspected genetic conditions, we were unable to quantify the contribution of CNVs to the diagnostic yield. One particular concern regarding the use of ES as a testing modality in renal patients is the effect of pseudogenes on the ability to reliably detect PKD1 variants.40 Due to this limitation, we did not include patients who had a clinical diagnosis of ADPKD. Despite these concerns, ES detected PKD1 variants in nine patients, highlighting the ability of ES to distinguish between cystic kidney disease phenocopies. In addition, we were unable to quantify the contribution to diagnostic yield of variable number tandem repeats (VNTRs) such as the MUC1 VNTR associated with medullary cystic renal disease, as VNTRs are challenging to identify through ES.41 Some of these technical limitations are likely be overcome in the future as genome sequencing becomes more widely established as the preferred testing modality in the clinical setting.
Although we collected a considerable amount of phenotypic data for each patient in the study, most of the clinical phenotype data was collected at study enrollment and not at onset of renal disease, meaning the age of onset of renal disease may be inaccurate, particularly in adult patients. For the same reasons, although we were able to see trends in diagnostic clinical groups, analyzing specific phenotypic features (such as hematuria and proteinuria) as clinical predictors of ES diagnosis was not possible. We also recognize the high diagnostic yield is due to selecting those patients who are most likely to have GKD as determined by MDT discussion, thereby limiting generalization to other service delivery models. Given the finite resources in most settings, the purpose of this study was to determine whether ES demonstrates diagnostic and clinical utility in selected patients, and to provide insight into clinical characteristics associated with higher yield to help clinicians prioritize patients for testing. Furthermore, careful assessment of patients and testing those who are more likely to benefit represents the current clinical approach in our health-care system. Finally, although we were able to determine immediate changes in clinical care following ES, long term clinical outcome data could not be captured in the study timeframe, and in most cases, these clinical decisions were at the discretion of the treating nephrologist.
Conclusions
ES identified a molecular diagnosis in almost 40% of patients referred for testing in a multidisciplinary RGC context. Our results confirm that in a pragmatic pediatric and adult cohort with suspected monogenic kidney disease, ES was not only valuable for establishing a specific molecular diagnosis, but also demonstrated substantial quantifiable impacts on clinical management. These findings, including the predictors of positive diagnosis, motivate further examination and validation in other cohorts. Further prospective studies evaluating the cost-effectiveness and implementation aspects of genomic testing in this population are required to improve access to testing for those most likely to benefit.
References
Thomas B, Matsushita K, Abate KH, et al. Global cardiovascular and renal outcomes of reduced GFR. J Am Soc Nephrol. 2017;28:2167–2179.
Groopman EE, Rasouly HM, Gharavi AG. Genomic medicine for kidney disease. Nat Rev Nephrol. 2018;14:83–104.
Groopman EE, Marasa M, Cameron-Christie S, et al. Diagnostic utility of exome sequencing for kidney disease. N Engl J Med. 2019;380:142–151.
Mann N, Braun DA, Amann K, et al. Whole-exome sequencing enables a precision medicine approach for kidney transplant recipients. J Am Soc Nephrol. 2019;30:201–215.
Vivante A, Hildebrandt F. Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol. 2016;12:133–146.
Rabbani B, Mahdieh N, Hosomichi K, Nakaoka H, Inoue I. Next-generation sequencing: impact of exome sequencing in characterizing Mendelian disorders. J Hum Genet. 2012;57:621–632.
Bresin E, Daina E, Noris M, et al. Outcome of renal transplantation in patients with non-Shiga toxin-associated hemolytic uremic syndrome: prognostic significance of genetic background. Clin J Am Soc Nephrol. 2006;1:88–99.
Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, et al. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2007;18:2392–2400.
Gross O, Licht C, Anders HJ, et al. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 2012;81:494–501.
Varner JD, Chryst-Stangl M, Esezobor CI, et al. Genetic testing for steroid-resistant-nephrotic syndrome in an outbred population. Front Pediatr. 2018;6:307.
Clark MM, Stark Z, Farnaes L, et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom Med. 2018;3:16.
Malinowski J, Miller DT, Demmer L, et al. Systematic evidence-based review: outcomes from exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability. Genet Med. 2020;22:986–1004.
Lata S, Marasa M, Li Y, et al. Whole-exome sequencing in adults with chronic kidney disease: a pilot study. Ann Intern Med. 2018;168:100–109.
Thomas CP, Freese ME, Ounda A, et al. Initial experience from a renal genetics clinic demonstrates a distinct role in patient management. Genet Med. 2020;22:1025–1035.
Warejko JK, Tan W, Daga A, et al. Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2018;13:53–62.
Mallett A, Fowles LF, McGaughran J, Healy H, Patel C. A multidisciplinary renal genetics clinic improves patient diagnosis. Med J Aust. 2016;204:58–59.
Alkanderi S, Yates LM, Johnson SA, Sayer JA. Lessons learned from a multidisciplinary renal genetics clinic. QJM. 2017;110:453–457.
von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. J Clin Epidemiol. 2008;61:344–349.
Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)-a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377–381.
Harris PA, Taylor R, Minor BL, et al. The REDCap consortium: building an international community of software platform partners. J Biomed Inform. 2019;95:103208.
Sadedin SP, Dashnow H, James PA, et al. Cpipe: a shared variant detection pipeline designed for diagnostic settings. Genome Med. 2015;7:68.
Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32:557–563.
Stark Z, Tan TY, Chong B, et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016;18:1090–1096.
Little MH, Quinlan C. Advances in our understanding of genetic kidney disease using kidney organoids. Pediatr Nephrol (Berlin, Germany). 2019;35:915–926.
Antonarakis SE, Beckmann JS. Mendelian disorders deserve more attention. Nat Rev Genet. 2006;7:277–282.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Rehm HL, Bale SJ, Bayrak-Toydemir P, et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med. 2013;15:733–747.
Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067.
Connaughton DM, Kennedy C, Shril S, et al. Monogenic causes of chronic kidney disease in adults. Kidney Int. 2019;95:914–928.
Mallett AJ, McCarthy HJ, Ho G, et al. Massively parallel sequencing and targeted exomes in familial kidney disease can diagnose underlying genetic disorders. Kidney Int. 2017;92:1493–1506.
Pei Y. Diagnostic approach in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2006;1:1108–1114.
Feneberg R, Schaefer F, Zieger B, Waldherr R, Mehls O, Scharer K. Percutaneous renal biopsy in children: a 27-year experience. Nephron. 1998;79:438–446.
Sinha MD, Lewis MA, Bradbury MG, Webb NJ. Percutaneous real-time ultrasound-guided renal biopsy by automated biopsy gun in children: safety and complications. J Nephrol. 2006;19:41–44.
Stark Z, Schofield D, Alam K, et al. Prospective comparison of the cost-effectiveness of clinical whole-exome sequencing with that of usual care overwhelmingly supports early use and reimbursement. Genet Med. 2017;19:867–874.
Wright CF, McRae JF, Clayton S, et al. Making new genetic diagnoses with old data: iterative reanalysis and reporting from genome-wide data in 1,133 families with developmental disorders. Genet Med. 2018;20:1216–1223.
White S, Jacobs C, Phillips J. Mainstreaming genetics and genomics: a systematic review of the barriers and facilitators for nurses and physicians in secondary and tertiary care. Genet Med. 2020;22:1149–1155.
Nestor JG, Marasa M, Milo-Rasouly H, et al. Pilot study of return of genetic results to patients in adult nephrology. Clin J Am Soc Nephrol. 2020;15:651–664.
Jayasinghe K, Quinlan C, Stark Z, et al. Renal genetics in Australia: kidney medicine in the genomic age. Nephrology (Carlton). 2018;24:279–286.
Lelieveld SH, Spielmann M, Mundlos S, Veltman JA, Gilissen C. Comparison of exome and genome sequencing technologies for the complete capture of protein-coding regions. Hum Mutat. 2015;36:815–822.
Mallawaarachchi AC, Hort Y, Cowley MJ, et al. Whole-genome sequencing overcomes pseudogene homology to diagnose autosomal dominant polycystic kidney disease. Eur J Hum Genet. 2016;24:1584–1590.
Kirby A, Gnirke A, Jaffe DB, et al. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet. 2013;45:299–303.
Acknowledgements
The study was funded by the Melbourne Genomics Health Alliance (Melbourne Genomics) and grants from the Royal Children’s Hospital Foundation and Australian Genomics Health Alliance (National Health and Medical Research Council APP1113531). Melbourne Genomics is funded by ten member organizations and the State Government of Victoria (Department of Health and Humans Services). The research conducted at the Murdoch Children’s Research Institute was supported by the Victorian Government’s Operational Infrastructure Support Program. K.J. was supported by the Royal Australian College of Physicians Jacquot Research Entry Scholarship and an Australian Government Research Training Program (RTP) Scholarship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jayasinghe, K., Stark, Z., Kerr, P.G. et al. Clinical impact of genomic testing in patients with suspected monogenic kidney disease. Genet Med 23, 183–191 (2021). https://doi.org/10.1038/s41436-020-00963-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-00963-4
Keywords
This article is cited by
-
What is the power of a genomic multidisciplinary team approach? A systematic review of implementation and sustainability
European Journal of Human Genetics (2024)
-
The emerging role of clinical genetics in pediatric patients with chronic kidney disease
Pediatric Nephrology (2024)
-
A step-by-step, multidisciplinary strategy to maximize the yield of genetic testing in pediatric patients with chronic kidney diseases
Pediatric Nephrology (2024)
-
Predictors of the utility of clinical exome sequencing as a first-tier genetic test in patients with Mendelian phenotypes: results from a referral center study on 603 consecutive cases
Human Genomics (2023)
-
ClinVar and HGMD genomic variant classification accuracy has improved over time, as measured by implied disease burden
Genome Medicine (2023)
