
Abstract
Purpose
To investigate if specific exon 38 or 39 KMT2D missense variants (MVs) cause a condition distinct from Kabuki syndrome type 1 (KS1).
Methods
Multiple individuals, with MVs in exons 38 or 39 of KMT2D that encode a highly conserved region of 54 amino acids flanked by Val3527 and Lys3583, were identified and phenotyped. Functional tests were performed to study their pathogenicity and understand the disease mechanism.
Results
The consistent clinical features of the affected individuals, from seven unrelated families, included choanal atresia, athelia or hypoplastic nipples, branchial sinus abnormalities, neck pits, lacrimal duct anomalies, hearing loss, external ear malformations, and thyroid abnormalities. None of the individuals had intellectual disability. The frequency of clinical features, objective software-based facial analysis metrics, and genome-wide peripheral blood DNA methylation patterns in these patients were significantly different from that of KS1. Circular dichroism spectroscopy indicated that these MVs perturb KMT2D secondary structure through an increased disordered to ɑ-helical transition.
Conclusion
KMT2D MVs located in a specific region spanning exons 38 and 39 and affecting highly conserved residues cause a novel multiple malformations syndrome distinct from KS1. Unlike KMT2D haploinsufficiency in KS1, these MVs likely result in disease through a dominant negative mechanism.
Similar content being viewed by others

INTRODUCTION
Diverse developmental phenotypes resulting from distinct variants in the same gene are being increasingly recognized, largely due to wider application of next-generation sequencing and multicenter collaborations.1 These discoveries provide unique biological insights into gene and protein functions, and are critical for appropriate medical management, and counseling of patients and families.
Defective histone lysine methylation and chromatin remodeling defects underlie several congenital malformation disorders.2KMT2D encodes a very large (593 kDa) protein that catalyzes the mono-, di-, and trimethylation of the lysine 4 on histone 3 (H3K4) in a multiprotein complex.3 The KMT2D protein contains multiple known domains; however, the structure and function of several regions of the KMT2D protein remain unresolved. KMT2D also has a high level of predicted disorder content (55%) with many regions of intrinsic disorder, which are typical features of nuclear proteins that regulate transcription and chromatin organization.4 Kabuki syndrome type 1 (KS1, MIM 147920) is an autosomal dominant condition caused by loss-of-function KMT2D (MIM 602113) variants.5 Over 700 KMT2D variants have been reported in the literature in individuals with KS1. Approximately 80% of germline KMT2D variants causative for KS1 are predicted to be protein truncating and are thought to result in functional haploinsufficiency due to nonsense-mediated decay.6 In rare instances, individuals with KS1 have also been described to have a whole-gene deletion.7 Germline KMT2D missense variants (MVs) causative for KS1 cluster in certain functional regions of the protein, such as in the PHD fingers, RING-type zinc fingers, the FYR-N domain and the SET domain6 (Fig. 1a–c). Notably, germline MVs in some highly conserved regions of the protein have not yet been described in individuals with KS1.6 KS1 is characterized by neonatal hypotonia, feeding difficulties during infancy and early childhood, postnatal growth deficiency, skeletal anomalies, immune dysfunction, endocrine abnormalities, and congenital malformations of the heart, kidney, and palate.8 Almost all individuals with KS1 show global developmental delay and intellectual disability. Facial dysmorphism is the most distinguishing feature of the condition and is typically characterized by interrupted eyebrows, long palpebral fissures, eversion of the lateral part of the lower eyelids, large cupped ears, short columella, bulbous nasal tip, and pillowed lower lip.8,9 Here, we describe clustering of heterozygous KMT2D MVs, in a highly conserved central region of the protein, as causative of a novel phenotype with multiple malformations that is clinically and epigenetically distinct from KS1.

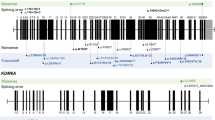
We studied multiple affected individuals from seven families with missense KMT2D variants restricted to a region that encodes for 54 amino acids flanked by Val3527 and Lys3583 (ENST00000301067.7; NM_003482.3). (a) Schematic representation of KMT2D exons with each alternating exon represented in dark or light red shade (introns are not depicted). (b) Frequency of KMT2D missense variants (from gnomAD) in the general population is shown in yellow. Deeper troughs represent higher frequency at that particular location. (c) Green lollipop graph denoting the missense KMT2D variants in individuals with Kabuki syndrome from the published literature.6 The y-axis in this graph represents the frequency of the variant in the published literature. The x-axis is a schematic for the protein denoting the location of important domains and regions of KMT2D. Note that variants identified in this study are located in parts of exons 38 and 39 with high missense constraint but without any variants in individuals with Kabuki syndrome. (d) The region of interest of the KMT2D gene and protein in more detail. The red horizontal bar shows parts of exons 38 (amino acid 3503–3580) and 39 (amino acid 3581–4510). The blue vertical bars denote the coiled-coil regions. Red lines indicate the location of the variants identified in this study. Pedigree of each family is shown under the corresponding variant. Standard symbols are used to denote affected (filled symbols) and unaffected (unfilled) individuals. All individuals who were tested but not found to carry familial KMT2D variants are denoted by “N”. Father in family 4 (F4; I:1) was found to be likely mosaic and is denoted by gray square. In this family, genetic testing was not possible for the first born child (F4; II:1) but is shown as affected based on the clinical history.
MATERIALS AND METHODS
Patient ascertainment, clinical characterization, and genetic studies
Ethical approval for molecular genetics research studies was obtained from the Research Ethics Committees of South Yorkshire (11/H1310/1), North West—Greater Manchester South (11/H1003/3), Cambridge South (10/H0305/83), the Republic of Ireland (GEN/284/12), Al-Ain Medical Human Research (ERH2015-3241-15-115), and East of England—Cambridge South (14/EE/1112). Informed consent for research studies was obtained from all participating families or individuals, as was permission to publish patient photographs. Ascertainment was driven by genotype in the first six families in whom KMT2D MVs of uncertain significance were identified by multigene panel testing or genome sequencing (GS) or exome sequencing (ES). The affected individual in family 7 was identified via targeted testing due to phenotypic similarity with other individuals described in this paper. All affected individuals underwent detailed clinical phenotyping (Table S1). Confirmation and segregation of allKMT2D variants was performed using conventional Sanger sequencing.
Total RNAs were extracted using RNeasy Mini kit (Qiagen) according to the manufacturer’s protocol and quantitative reverse transcription polymerase chain reaction (RT-qPCR) was performed using standard methods.
Facial analysis
Ten previously unpublished photographs at different ages of six individuals in Ex38/39 KMT2D MVs cohort were uploaded onto and the results were obtained using the Face2Gene RESEARCH application (FDNA Inc., Boston, MA) for objective analysis of facial dysmorphology. Comparison was made with two groups of photographs of ten individuals, each with molecularly confirmed KS1 or CHARGE syndrome (OMIM 214800). The sensitivity and specificity of this classification system was analyzed by receiver operating characteristic (ROC) curves, and the corresponding area under the curve (AUC) and p values were calculated as described previously.10
Peripheral blood DNA methylation
Peripheral blood was collected in EDTA tubes and genomic DNA was isolated using QIAamp DNA blood mini kit (Qiagen). Unmethylated cytosines were converted to uracil through bisulfite conversion using Zymo EZ DNA Methylation kit (Zymo Research). Genome-wide methylation was studied using Infinium MethylationEPIC BeadChip Kit (Illumina) (hereafter referred to as EPIC array) according to the manufacturer’s protocol. The array was stained and scanned using the Illumina iScan System. Data were processed using standard methods (Supplementary methods). Due to small number of samples and differences in ages of our patients and controls, we removed known age-associated probes.11,12,13 Principal component analysis (PCA) was used to visualize clustering of the samples based on differentially methylated positions (DMPs) between groups (p < 0.001). Overlap of DMPs between groups was assessed using InteractiVenn web based tool.14 Gene ontologies associated with DMPs were assessed using canonical pathway analysis (Ingenuity pathway analysis software, Qiagen, San Francisco, CA). Differentially methylated regions of the chromosome (DMRs) (>7 annotated CpGs) were analyzed on EPIC array data using the Bumphunter method15 in the Bioconductor package ChAMP.16
H3K4me3 methylation
The pFlag-CMV2 FUSION–KMT2D construct17 was modified by inserting sequences for KMT2D that encode residues 3387–3697. Expression plasmids harboring KMT2D missense variants were generated by site-directed mutagenesis according to standard methods. HEK-293T cells were cultured in Dulbecco’s Modified Eagle Medium with 10% fetal bovine serum, penicillin (100 U/ml) and streptomycin (100 mg/ml) (Life Technologies). HEK-293T cells were transiently transfected using the polyethylenimine method, following published protocols.18 Cells were harvested 48 hours after transfection and used for protein extraction and histone methyltransferase assay. HEK-293T cells were plated in 12-well culture dishes at a density of 2.5 × 104 cells/ml and then FLAG-KMT2D wild-type or mutated constructs were cotransfected together with an epigenetic reporter allele (“methyl reader”).19 Forty-eight hours after transfection, cells were washed and resuspended in 100 μl phosphate buffered saline (PBS) and subsequently plated into 96-well plates with black flat bottom (Corning®). The fluorescence signal was monitored using a Glomax 96 microplate luminometer (blue filter, Ex = 475 nm; Em = 500–550 nm) and was normalized to the methyl reader signal. Three or more biological replicates were performed for all assays.
Protein analysis
Coiled-coil domains were predicted by MARCOIL, a hidden Markov model–based program that predicts the existence and location of potential coiled-coil domains in protein sequences.20 MultiCoil was used for predicting two- and three-stranded coiled coils.21 Gene synthesis of the wild-type and mutant sequences for KMT2D that encode residues 3231–3600 or 3503–3600 was carried out by GENEWIZ Inc. (South Plainfield, NJ). The sequences were subcloned into the bacterial expression vector pOPINF22 with a polyhistidine (6xHis) tag. Standard methods were followed for optimization of soluble recombinant protein expression at 12 °C in E. coli “ArcticExpress” cells (Agilent Technologies) and affinity purification with nickel-charged affinity resin columns (Ni-NTA Agarose) using fast protein liquid chromatography (ÄKTA Chromatography System, GE Healthcare Life Sciences). Protein purity was assessed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting. Purified proteins were buffer exchanged into 100 mM phosphate buffer, pH 7.2 (68.4 mM Na2HPO4, 31.6 mM NaH2PO4) and analyzed by circular dichroism (CD) spectroscopy, using a Chirascan Plus (Applied Photophysics) spectropolarimeter. Analyses of experimental protein CD spectra were by DichroWeb.23
RESULTS
Genetic studies
We ascertained multiple individuals from seven families with missense variants restricted to a highly conserved region of 54 amino acids flanked by Val3527 and Lys3583 (ENST00000301067.7; NM_003482.3) (Figs. 1d and 2a, b; Fig. S1) (Supplementary case reports) (hereafter referred to as Ex38/39 KMT2D MVs). Identical de novo heterozygous exon 38 KMT2D c.10582C>G p.(Leu3528Val) variants was identified in probands of families 1 and 2. A de novo heterozygous exon 38 KMT2D c.10625T>C p.(Leu3542Pro) variant was identified in the proband of family 3. In family 4 (previously reported by Al-Gazali et al.24) and family 5 an identical heterozygous exon 38 KMT2D c.1658G>T p.(Gly3553Val) variant was seen in all affected individuals available for testing. In family 4, where multiple children were affected, the variant was shown to be mosaic in their father (Fig. S2) and in family 5 the variant had arisen de novo in the proband. In family 6, a heterozygous exon 39 KMT2D c.10745G>A p.(Arg3582Gln) variant was detected in the proband and his similarly affected mother. A de novo heterozygous exon 39KMT2D c.10744C>T p.(Arg3582Trp) variant, affecting the same residue that was substituted in family 6, was detected in family 7. Additional rare de novo or biallelic variants identified in our studies were all classed as likely benign (Table S2). All KMT2D MVs affected highly conserved residues (Fig. S1) and were absent in gnomAD, [Karczewski KJ, Francioli LC, Tiao G, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function Q7 intolerance across human protein-coding genes. 2019] dbSNP,25 and ClinVar.26 The c.10744C>T (COSM431206) and c.10745G>A (COSM4604032) variants were found in COSMIC27 (Table S1). Overall, these results were highly indicative that the variants were pathogenic.

(a) Photographs of individuals described here with missense KMT2D variants. Note the wide range of facial features. P2, proband from family 2, is shown at two different ages. Note facial asymmetry, hypertelorism, bilateral epicanthic folds, bulbous nasal tip, downturned corners of the mouth, microtia, and hypoplastic nipples. P3, proband from family 3, has a box-shaped head, bilateral microphthalmia, severely hypoplastic left pinna, and ectopic left external auditory canal. P5 and P7, probands from families 5 and 7 respectively, have prominent forehead, broad nasal root, flat midface, and thin upper lip. P7’s eyebrows are laterally flared. One individual with Kabuki syndrome type 1 (KS1) is shown for comparison. Note arched eyebrows, long palpebral fissures, eversion of the lateral part of the lower eyelids, large cupped ears, short columella, bulbous nasal tip, and pillowed lower lip. In individual with CHARGE syndrome note hypertelorism, bulbous and large nasal tip, and a repaired cleft lip. (b) Computerized tomography (CT) (i and ii) and T2-weighted magnetic resonance imaging (MRI) (iii) of P2 demonstrating absence of the posterior part of the semicircular canals (red arrows) and normal anatomy of the lateral and anterior semicircular canals. Brain MRI of P3 (iv) to show well-formed right and left middle ear cavities, with the right cavity being smaller than the left. Bilaterally the cochlea, semicircular canals, and inner auditory canals appear normal. T1-weighted MRI to demonstrate a small left optic globe with ballooning of the optic disc bilaterally (red arrows) and optic disc colobomata (v). CT imaging of P5 (vi, vii) demonstrating the presence of cysts in the lower jaw (red arrows). (c) Face2Gene analysis with confusion matrix showing that the system is able to predict correctly each group with a mean accuracy of 75.44%. (d) Receiver operating characteristic (ROC) graphs show the probability curve where the area under the curve (AUC) (0–1) represents the measure of separability between two groups. Score distributions show the distribution of those probabilities. The higher the AUC, the better the model is at distinguishing between two groups. (e) Principal component analysis (PCA) shows the four groups analyzed in the DNA methylation array clustering separately (p < 0.001). Importantly, the samples from individuals described in this study cluster together and separate from those with type 1 Kabuki syndrome. MV missense variant.
Phenotype and facial analyses
As none of the patients were clinically diagnosed with KS, we compiled the clinical features of all affected individuals in detail (Fig. 2a, b) (Table 1). The most consistent clinical features of the affected individuals identified here included choanal atresia, athelia or hypoplastic nipples, branchial sinus abnormalities, neck pits, abnormalities of the lacrimal ducts, hearing loss, external ear malformations, and thyroid abnormalities or functional thyroid disease.
To examine if the phenotype of the affected individuals is distinct from that of KS1, we compared the frequencies of major phenotypes observed in Ex38/39 KMT2D MVs cohort and in individuals with KS1 (Table 2). Only hearing loss and external ear abnormalities were consistent major features of both conditions. In contrast, cleft lip or palate, renal structural abnormalities, and seizures are common in KS1 but were absent in the Ex38/39 KMT2D MVs cohort. Due to the clinical overlap, we also compared the frequencies of phenotypes observed in the Ex38/39 KMT2D MVs cohort with individuals with CHARGE syndrome. This comparison also showed several differences (Table 2). External ear malformations, although seen in all three groups are of different nature (small or absent external ears in the Ex38/39 KMT2D MVs cohort, large prominent ears in KS1, and simple dysplastic in CHARGE syndrome). Markedly, intellectual disability that is almost universal in KS1 and CHARGE syndromes was absent in the Ex38/39 KMT2D MVs cohort. As facial dysmorphism is the most specific and sensitive feature of KS1,8,9 we performed computer-based objective analyses based on syndrome-specific classifiers of facial features. This showed that the facial features of individuals in the Ex38/39 KMT2D MVs cohort were significantly different to those with KS1 (AUC = 0.918; p = 0.018) and CHARGE syndrome (AUC = 0.902; p = 0.025) (Fig. 2c, d). Overall, these analyses were highly suggestive that the phenotype of the individuals described in this study is distinct from that of Kabuki and CHARGE syndromes.
Peripheral blood DNA methylation
Since DNA methylation signatures can differentiate pathogenic and nonpathogenic KMT2D variants,28 we examined if the epigenetic profile of the condition we described herein is distinct from that of KS1. We compared the DNA methylation patterns in peripheral blood collected from the following individuals: four affected individuals from the Ex38/39 KMT2D MVs cohort, four individuals with KS1, four with CHARGE syndrome, and four controls (Table S3). We validated our results by comparing them with the hypo- and hypermethylated CpG sites in individuals with KS1 listed by Butcher et al. (Fig. S3).28 The PCA analysis of the DNA methylation profiles of samples from the Ex38/39 KMT2D MVs cohort clustered together and differed from those of controls, KS1, and CHARGE syndrome (Fig. 2e). Intersection analysis showed that the samples from the Ex38/39 KMT2D MVs cohort and KS1 share only 915 differentially methylated CpG sites (p < 0.001) (Fig. S4). These results were highly suggestive that Ex38/39 KMT2D MVs result in a condition that is epigenetically distinct from both Kabuki and CHARGE syndromes.
RT-qPCR
To explore the disease mechanism we studied the effect of these variants on the expression of KMT2D. Fibroblasts were obtained from two affected individuals from our cohort (proband from family 7 and family 3) (Table S3) and one control. Using RT-qPCR, we observed no significant differences in the level of KMT2D expression in fibroblasts of affected individuals compared with the control (Fig. S5).
Histone H3K4 methylation assays
To experimentally test the functional impact of KMT2D missense variants, we generated FLAG-tagged versions of six KMT2D missense alleles: p.Leu3528Val, p.Leu3542Pro, p.Gly3553Val, p.Gln3575His,29 p.Arg3582Trp, and p.Arg3582Gln. Fluorescence data revealed no changes in trimethylation H3K4 levels between all the missense variants tested in this assay compared withKMT2D normal control activity (Fig. S6).
Protein analysis
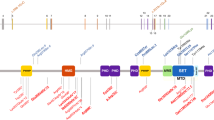
As the variants did not appear to impact on KMT2D expression or direct H3K4 trimethylation activity, we considered the possibility that variants affected protein–protein interactions with KMT2D. The variants identified in this study do not occur in a known functional domain of the protein, but are instead in a central, highly conserved region that also contains short polyQ tracts (Fig. S1). All of the variants occur within or close to a coiled-coil domain (residues 3562–3614; Fig. 3a) predicted by MARCOIL.20 Furthermore, MultiCoil21 identified heptad repeat sequences that mediate a possible coiled-coil dimer between residues 3506 and 3610 (heptad net plot, Fig. 3b). Analysis of the CD spectrum of the KMT2D wild-type protein fragment (3503–3600 residues, molecular weight 11.8 kDa) by DichroWeb23 revealed the following predicted proportions of secondary structure: 0.14 α-helix, 0.26 β-strand, 0.27 turn, and 0.32 disordered. A larger KMT2D wild-type protein fragment (3231–3600 residues, molecular weight 40.7 kDa) had the following proportions: 0.22 α-helix, 0.18 β-strand, 0.19 turn, and 0.41 disordered (Fig. S7). All recombinant KMT2D proteins with missense variants had perturbed secondary structure with decreased disorder and increased proportions of predicted α-helical structure (Fig. 3c).

(a) Central, highly conserved region of KMT2D containing a coiled-coil domains predicted by MARCOIL (blue trace). All KMT2D missense variants described here (red lines) occur within or close to the predicted coiled-coil domain (residues 3562–3614). KMT2D fusion proteins (residues 3503–3600; gray bar) contained the missense variants described here. (b) Heptad net plot view of a potential coiled-coil domain, predicted by MARCOIL between residues 3511 and 3559 of KMT2D, showing approximately seven heptad repeats of hydrophobic or nonpolar residues (black letters) and charged residues (blue or red letters). Missense variants of residues within or close to the predicted coiled-coil domain are indicated by red boxes. Positions of residues within the predicted heptad repeat sequences are labeled a to g. Residues at the “a” and “d” positions (gray boxes), which include Leu3528 and Leu3542, form the hydrophobic seam in a coiled coil. (c) Upper panel: expression of wild-type and mutant recombinant KMT2D fusion proteins, as indicated. Lower panel: circular dichroism (CD) spectroscopy traces of recombinant KMT2D wild-type and mutant proteins showing moderate levels of disordered secondary structure in the wild-type protein (black trace) with perturbed secondary structure and higher proportions of ɑ-helical structure in all recombinant KMT2D proteins with missense variants (purple, red, light blue, and green traces).
Differentially methylated region analysis
We used the data generated from our peripheral blood DNA methylation experiments to explore the likely dysregulated pathways. Within the 5162 differentially methylated CpG sites in Ex38/39 KMT2D MVs cohort compared with control samples, we observed significant enrichment for CpG sites corresponding to genes related to morphology of the head, embryonic development, cell proliferation, and development of body axis (Fig. S4). Next, we investigated the potential disease mechanism by analyzing DMRs in the Ex38/39 KMT2D MVs cohort compared with control samples. We identified 83 such DMRs including several genes associated with clinical features, such as hearing impairment or hypothyroidism, that we observed in the Ex38/39 KMT2D MVs cohort (Table S4) (Fig. S4).
DISCUSSION
Multiple lines of evidence suggest that the Ex38/39 KMT2D MVs identified in this study are the cause of the phenotypes of the affected individuals. These include (1) the de novo occurrence of the variants in five families (Fig. 1), (2) segregation of the variants with the phenotype in a multiplex family (family 6), (3) presence of variant in mosaic state in an unaffected parent of another multiplex family (family 4), (4) absence of these variants from control databases, (5) identical variants in families 1 and 2 (p.Leu3528Val) and in families 4 and 5 (p.Glu3553Val), (6) distinct variants affecting the same codon in families 6 and 7 (p.Arg3582Glu) and (p.Arg3582Trp), (7) clustering of the variants within a span of just 54 amino acids in a protein of size 5537 amino acids with very strong evolutionary conservation of all affected residues (Fig. S1), (8) strong similarity of the phenotype in families 1–6 that were ascertained on the basis of genotype, and (9) identification of the variant in family 7 through targeted single-gene phenotype-led testing (Table 1 and S1) (Fig. 2).
The affected individuals we describe share some features with KS1, including hearing loss, dental anomalies, feeding difficulties, failure to thrive, and abnormalities of the immune system. However, the most commonly observed malformations in individuals with KS1, including cleft palate, congenital heart disease, and renal anomalies, were infrequent or absent in the Ex38/39 KMT2D MVs cohort (Table 2).8 Importantly, the affected individuals in our study did not display intellectual disability or hypotonia, which are almost universal features in all patients with KS1. Speech delay noted in several individuals was attributable to their hearing impairment. Furthermore, the typical dysmorphic features of KS1 were not seen in the cohort described here.8 This was supported by the results of our objective facial analysis studies (Fig. 2). We found that the DNA methylation pattern of individuals described here was distinct from what has been described in KS1. Notably, the DNA methylation studies were performed in a small number of individuals and testing in a larger patient cohort will be required to validate our findings. Nevertheless, collectively these results strongly suggest that the restricted spectrum of KMT2D missense variants in exon 38 and 39 identified in this study result in a multiple malformation disorder that is genetically, phenotypically, and epigenetically distinct from KS1.
The variants described here are flanked by Val3527 and Lys3583. However, it remains to be seen if variants outside this window may also result in the same phenotype. Furthermore, the phenotype spectrum of individuals described here is very wide, ranging from early death of three individuals (from families 3 and 4) to a relatively mild phenotype (e.g., the affected mother in family 6). This may be due to allelic heterogeneity or other genetic, epigenetic, or environmental factors. The identification of larger cohorts of individuals will be required to resolve these questions.
Some features of the Ex38/39 KMT2D MVs cohort overlap with those of CHARGE syndrome (OMIM 214800), including coloboma, choanal atresia, and ear anomalies (Table S5).30 However, none of the affected individuals fulfill the clinical diagnostic criteria for CHARGE syndrome.30 Molecularly, KMT2D and CHD7 both interact with members of the WAR complex31 and clinical overlap between KMT2D variants and CHARGE syndrome has been previously postulated.29,31 Two of these previous reports described individuals with clinical features that match those of the Ex38/39 KMT2D MVs cohort and that carry KMT2D missense variants, one with p.Gln3575His and another with p.Leu3564Val, that lie within the central conserved KMT2D region we describe.29,32 We propose that these previously published individuals have neither KS1 nor CHARGE syndrome, and fit better with the condition that we describe in this study. Other disorders that phenotypically overlap with the condition described here include branchiootorenal syndrome (BORS, OMIM 113650), branchiootic syndrome 2 (OMIM 120502), branchiooculofacial syndrome (OMIM 113620), and Bamforth–Lazarus syndrome (OMIM 241850) (Table S5).
Athelia or hypoplasia of the nipples is a rare clinical feature of unknown incidence and was present in more than half the patients in our report. On the contrary, most individuals with KS1 tend to develop prominent breasts during infancy and early childhood. A single case report has described an association between hypoplastic nipples and CHARGE syndrome.33 The combination of athelia and choanal atresia is an even rarer association, previously only described in carbimazole embryopathy.34 In the absence of an antenatal history of maternal carbimazole use, this combination of features would be very suggestive of the condition described in this paper. Of note, carbimazole is a pro-drug that is converted to methimazole, which inhibits thyroid peroxidase. We speculate that theKMT2D variants described here perhaps directly or indirectly result in dysfunction, downregulation, or inhibition of thyroid peroxidase or developmental pathways involving this enzyme. On review of the literature with a focus on the combination of choanal atresia and athelia, we identified two previous case reports describing features very similar to the families described here including athelia, choanal atresia, failure to thrive, congenital cardiac disease, and preauricular pits.35,36 Due to the clinical similarities, we suspect that our report provides a possible explanation for the etiology of disease in these historical cases.
In contrast to the observations for individuals with Kabuki-causingKMT2D missense variants,17 the H3K4 methyltransferase activities of the variants described here were normal (Fig. S6). Notably, KMT2D also possesses H3K4 me1 and me2 methyltransferase activity that we have not tested. However, given the genetic and clinical findings, it appears unlikely that the phenotype results simply from the loss of H3K4 methyltransferase function. Importantly, the variants identified in this study are in a highly conserved central region that contains predicted coiled-coil domains (Fig. 1b) as well as short polyQ tracts (Fig. S1). The polyQ tracts at the C-terminus end of the highly conserved central region are likely to be unstructured and to destabilize coiled-coil formation, potentially with the formation of intrinsically disordered regions of secondary protein structure. The results of our CD experiments (Fig. 3) suggest that the central region of KMT2D is indeed disordered, which is consistent with the usual major roles of intrinsically disordered regions in mediating and modulating protein–protein interactions.4,37 However, pathogenic missense variants in the central region, as in the Ex38/39 KMT2D MVs cohort, could prevent normal transitions between disordered and ɑ-helical structure in a coiled-coil domain, or reduce the levels of intrinsic disorder, thereby disrupting normal specific protein–protein interactions in the KMT2D chromatin remodeling complex.
In the peripheral blood of affected individuals one of the most significant hypomethylated DMRs that we observed encompasses the transcription start site (TSS) region of HOXA2 (OMIM 604685), variants in which cause autosomal dominant microtia with or without hearing impairment (612290) (Fig. S4). HOXA2 is a fundamental transcriptional factor that regulates EYA1 (linked with BORS) and orchestrates morphogenesis of the auricle. Notably, ear anomalies were seen very frequently in the Ex38/39 KMT2D MVs cohort. We also identified hypermethylation in the gene body of PAX8 (OMIM 167415), variants in which cause congenital hypothyroidism due to thyroid dysgenesis or hypoplasia (OMIM 218700) (Fig. S4). Hypothyroidism was a frequent feature in the Ex38/39 KMT2D MVs cohort. Furthermore, a few CpG sites corresponding to genes involved in neural crest development showed altered DNA methylation pattern. In particular, we observed hypomethylation in the regions corresponding to ZIC3 (OMIM 300205), TFAP2E (OMIM 614428), and SOX10 (OMIM 602229) and a hypermethylation in the region corresponding to ZEB2 (OMIM 605802). All these regulators influence neural crest migration.38 Although the expression of these genes requires to be tested in relevant tissue, our results suggest that the variants described in this study might be deleterious to the normal expression or function of key transcription factors that might impair specific developmental processes.
In summary, we describe a variable disorder caused by missense variants in parts of exons 38 and 39 of KMT2D that is clinically, genetically, and epigenetically distinct from KS1. This condition is characterized by anomalies of the branchial arch, ears, choanae, lacrimal ducts, nipples, thyroid, growth, and the immune system. This work provides insights into the diverse roles of KMT2D in embryonic development and uncovers the importance of a previously unstudied region of this critical protein that is likely to be an intrinsically disordered region that mediates and modulates protein–protein interactions. Further work will be required to determine the link between theKMT2D variants described here and the human and cellular phenotypes observed. Overall, this work emphasizes the possibility of undiscovered phenotypes linked with even well-studied genes.39,40
Change history
23 March 2020
An incorrect reference was cited in the fourth sentence of the Results section. The correct citation should have been Al-Gazali LI, Hamid Z, Hertecant J et al. An autosomal recessive syndrome of choanal atresia, hypothelia/athelia and thyroid gland anomalies overlapping Bamforth syndrome, ANOTHER syndrome and methimazole embryopathy. Clin Dysmorphol 2002;2:79–85. This has now been corrected in both the PDF and HTML versions of the Article.
References
Reijnders MRF, Ansor NM, Kousi M, et al. RAC1 missense mutations in developmental disorders with diverse phenotypes. Am J Hum Genet. 2017;101:466–477.
Faundes V, Newman WG, Bernardini L, et al. Histone lysine methylases and demethylases in the landscape of human developmental disorders. Am J Hum Genet. 2018;102:175–187.
Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012;81:65–95.
Staby L, O’Shea C, Willemoes M, Theisen F, Kragelund BB, Skriver K. Eukaryotic transcription factors: paradigms of protein intrinsic disorder. Biochem J. 2017;474:2509–2532.
Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–793.
Faundes V, Malone G, Newman WG, Banka S. A comparative analysis of KMT2D missense variants in Kabuki syndrome, cancers and the general population. J Hum Genet. 2019;64:161–170.
Banka S, Howard E, Bunstone S, et al. MLL2 mosaic mutations and intragenic deletion-duplications in patients with Kabuki syndrome. Clin Genet. 2013;83:467–471.
Adam MP, Banka S, Bjornsson HT, et al. Kabuki syndrome: international consensus diagnostic criteria. J Med Genet. 2019;56:89–95.
Banka S, Veeramachaneni R, Reardon W, et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur J Hum Genet. 2012;20:381–388.
Gurovich Y, Hanani Y, Bar O, et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat Med. 2019;25:60–64.
Alisch RS, Barwick BG, Chopra P, et al. Age-associated DNA methylation in pediatric populations. Genome Res. 2012;22:623–632.
Hannum G, Guinney J, Zhao L, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–367.
Perez RF, Santamarina P, Tejedor JR, et al. Longitudinal genome-wide DNA methylation analysis uncovers persistent early-life DNA methylation changes. J Transl Med. 2019;17:15.
Heberle H, Meirelles GV, da Silva FR, Telles GP, Minghim R. InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics. 2015;16:169.
Jaffe AE, Murakami P, Lee H, et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int J Epidemiol. 2012;41:200–209.
Morris TJ, Butcher LM, Feber A, et al. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics. 2014;30:428–430.
Cocciadiferro D, Augello B, De Nittis P, et al. Dissecting KMT2D missense mutations in Kabuki syndrome patients. Hum Mol Genet. 2018;27:3651–3668.
Thomas M, Lu JJ, Ge Q, Zhang C, Chen J, Klibanov AM. Full deacylation of polyethylenimine dramatically boosts its gene delivery efficiency and specificity to mouse lung. Proc Natl Acad Sci USA. 2005;102:5679–5684.
Bjornsson HT, Benjamin JS, Zhang L, et al. Histone deacetylase inhibition rescues structural and functional brain deficits in a mouse model of Kabuki syndrome. Sci Transl Med. 2014;6:256ra135.
Delorenzi M, Speed T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics. 2002;18:617–625.
Wolf E, Kim PS, Berger B. MultiCoil: a program for predicting two- and three-stranded coiled coils. Protein Sci. 1997;6:1179–1189.
Berrow NS, Alderton D, Sainsbury S, et al. A versatile ligation-independent cloning method suitable for high-throughput expression screening applications. Nucleic Acids Res. 2007;35:e45.
Whitmore L, Wallace BA. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32(Web Server issue):W668–W673.
Gazali et al. Clinical Dysmorphology 2002. https://journals.lww.com/clindysmorphol/Fulltext/2002/04000/An_autosomal_recessive_syndrome_of_choanal.1.aspx.
Smigielski EM, Sirotkin K, Ward M, Sherry ST. dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res. 2000;28:352–355
Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–D1067.
Forbes SA, Beare D, Boutselakis H, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45(D1):D777–D783.
Butcher DT, Cytrynbaum C, Turinsky AL, et al. CHARGE and Kabuki syndromes: gene-specific DNA methylation signatures identify epigenetic mechanisms linking these clinically overlapping conditions. Am J Hum Genet. 2017;100:773–788.
Badalato L, Farhan SM, Dilliott AA, et al. KMT2D p.Gln3575His segregating in a family with autosomal dominant choanal atresia strengthens the Kabuki/CHARGE connection. Am J Med Genet A. 2017;173:183–189.
Trider CL, Arra-Robar A, van Ravenswaaij-Arts C, Blake K. Developing a CHARGE syndrome checklist: health supervision across the lifespan (from head to toe). Am J Med Genet A. 2017;173:684–691.
Schulz Y, Freese L, Manz J, et al. CHARGE and Kabuki syndromes: a phenotypic and molecular link. Hum Mol Genet. 2014;23:4396–4405.
Sakata S, Okada S, Aoyama K, et al. Individual clinically diagnosed with CHARGE syndrome but with a mutation in KMT2D, a gene associated with Kabuki syndrome: a case report. Front Genet. 2017;8:210.
Glustein JZ, Weill M, Steinberg A. Anomalies of the nipple: an additional finding in CHARGE syndrome. Am J Med Genet. 1996;61:201.
Foulds N, Walpole I, Elmslie F, Mansour S. Carbimazole embryopathy: an emerging phenotype. Am J Med Genet A. 2005;132A:130–135.
Hisama FM, Reyes-Mugica M, Wargowski DS, Thompson KJ, Mahoney MJ. Renal tubular dysgenesis, absent nipples, and multiple malformations in three brothers: a new, lethal syndrome. Am J Med Genet. 1998;80:335–342.
Horvath GA, Armstrong L. Report of a fourth individual with a lethal syndrome of choanal atresia, athelia, evidence of renal tubulopathy, and family history of neck cysts. Am J Med Genet A. 2007;143A:1231–1235.
Tompa P, Schad E, Tantos A, Kalmar L. Intrinsically disordered proteins: emerging interaction specialists. Curr Opin Struct Biol. 2015;35:49–59.
Simoes-Costa M, Bronner ME. Establishing neural crest identity: a gene regulatory recipe. Development. 2015;142:242–257.
Banka S, Sayer R, Breen C, et al. Genotype-phenotype specificity in Menke-Hennekam syndrome caused by missense variants in exon 30 or 31 of CREBBP. Am J Med Genet A. 2019;179:1058–1062.
Cuvertino S, Stuart HM, Chandler KE. et al. ACTB loss-of-function mutations result in a pleiotropic developmental disorder. Am J Hum Genet. 2017;101:1021–1033.
Acknowledgements
We are grateful to all the individuals and their families for taking part in the study. S.C. and S.B. acknowledge the support of Newlife Charity (grant number 16–17/10). This work was also supported by Chile’s National Commission for Scientific and Technological Research (CONICYT PhD studentship 72160007 to V.F.), a Wellcome Trust Strategic Award (102627/Z/13/Z to R.T. and D.v.H.), the British Heart Foundation (Clinical Training fellowship FS/13/32/30069 to V.H.), a Sir Jules Thorn Award for Biomedical Research (JTA/09 to E.S. and C.A.J), a Wellcome Trust Vacation Scholarship (213312/Z/18/Z to A.C.), and the Telethon—Italy (grant number GGP13231 to G.M.). The Protein Production and Circular Dichroism Spectroscopy Facilities at the University of Leeds are supported by the Royal Society Wolfson Laboratory Refurbishment scheme and the Wellcome Trust (062164/Z/00/Z). This work was supported by the National Institute for Health Research (NIHR) Manchester Biomedical Research Centre. The Deciphering Developmental Disorders (DDD) study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009–003). This study makes use of DECIPHER (http://decipher.sanger.ac.uk), which is funded by the Wellcome. See Nature PMID 25533962 or www.ddduk.org/access.html for full acknowledgement. Part of the data presented here was provided through access to the data and findings generated by the 100,000 Genomes Project, which is funded by the NIHR and NHS England (full acknowledgement on https://www.genomicsengland.co.uk/about-gecip/publications/). We thank Hans T. Bjornsson (McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University) for his kind gift of the epigenetic reporter allele (methyl reader).
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Web resources
www.agilent.com/genomics/suredesign
https://app.face2gene.com/research
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cuvertino, S., Hartill, V., Colyer, A. et al. A restricted spectrum of missense KMT2D variants cause a multiple malformations disorder distinct from Kabuki syndrome. Genet Med 22, 867–877 (2020). https://doi.org/10.1038/s41436-019-0743-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0743-3
