
Abstract
Purpose
To date, there has not been a large, systematic evaluation of the prevalence of germline risk variants in urothelial carcinoma (UC).
Methods
We evaluated the frequency of germline pathogenic and likely pathogenic variants in 1038 patients with high-risk UC who underwent targeted clinical germline testing. Case–control enrichment analysis was performed to screen for pathogenic variant enrichment in 17 DNA repair genes in 1038 UC patients relative to cancer-free individuals.
Results
Among 1038 patients with UC, the cumulative frequency of patients with pathogenic variants was 24%; 18.6% of patients harbored ≥1 actionable germline variant with preventive or therapeutic utility. MSH2 (34/969, 3.5%) and BRCA1/2 (38/867, 4.4%) germline variants had the highest frequency. Germline variants in DNA damage repair genes accounted for 78% of pathogenic germline variants. Compared to the cancer-free cohort, UC patients had significant variant enrichment in MSH2 (odds ratio [OR]: 15.4, 95% confidence interval [CI]: 7.1–32.7, p < 0.0001), MLH1 (OR: 15.9, 95% CI: 4.4–67.7, p < 0.0001), BRCA2 (OR: 5.7, 95% CI: 3.2–9.6, p < 0.0001), and ATM (OR: 3.8, 95% CI: 1.8–8.3, p = 0.02).
Conclusion
In this study, 24% of UC patients harbored pathogenic germline variants and 18.6% had clinically actionable variants. MLH1 and MSH2 were validated as UC risk genes while ATM and BRCA2 were highlighted as potential UC predisposition genes. This work emphasizes the utility of germline testing in selected high-risk UC cohorts.
Similar content being viewed by others
Introduction
The burden of bladder cancer in the United States is substantial with over 80,000 new cases and 17,000 deaths expected in 2019.1 Despite the recent incremental therapeutic advances in the treatment of urothelial carcinoma (UC), primary and secondary prevention strategies may have the greatest influence on reducing morbidity and mortality of this disease. As such, in-depth understanding of the environmental and genetic contributors to increased risk of bladder cancer is warranted.
While previous studies have demonstrated familial risk and heritability for bladder cancer,2,3 prognostic and therapeutic implications of germline variants in bladder cancer patients are increasingly recognized.4,5 UCs are known component cancers of Lynch syndrome, which is caused by pathogenic variants in mismatch repair genes (including MLH1, MSH2, MSH6, and PMS2).6,7,8 In fact, the cumulative risk of developing bladder cancer by age 70 years in MSH2-associated Lynch syndrome patients was reported to be 12.3% for men and 2.6% for women.8 In addition, a recent study of 98 Chinese patients with bladder cancer reported germline DNA damage repair gene (DRG) variants in 10.2% of cases.5 However, currently germline data in UC is limited to small studies in specific ethnic groups or studies with incomplete family histories. These limitations may stem from poor access to clinical genetics evaluation, lack of awareness of the potential value of germline testing in UC patients, cost of testing, or availability of germline DNA biobanking. Collectively, the lack of available data has made it difficult to endorse a role for germline genetic testing in clinical practice guidelines, and the absence of practice guidelines then limits the availability of data on which to base recommendations. The lack of published data underlies the need for a larger systematic evaluation of germline variants in UC.
Herein, we describe the prevalence of germline cancer risk variants among a large cohort of high-risk UC patients completing genetic testing. This study is the largest to date to examine germline cancer risk variants and identify associated clinical factors in patients with UC.
MATERIALS AND METHODS
Patient cohort
Personal history information from requisition forms and medical records was manually reviewed for 1038 patients with urothelial carcinoma who completed germline genetic testing via one commercial laboratory between 2015 and 2018. Clinical germline genetic testing was performed at Invitae (San Francisco, CA), a laboratory that is certified for clinical use and patient reporting under CLIA and accredited by the College of American Pathologists (CAP). Each patient had between 1 and 130 genes analyzed. The majority of patients (n = 1002, 97%) completed a multigene panel test of ≥5 genes. Patient data were de-identified and Western Institutional Review Board provided study oversight and approval. Western Institutional Review Board protocol number 1167406 waived the requirement to obtain written patient informed consent. Ethnicity data were collected for most patients by self-report at the time of test ordering and grouped based on categories reported in the Exome Aggregation Consortium (ExAC)9 database.
Targeted sequencing
Genomic DNA was extracted from whole blood using a QiaSymphony (Qiagen, Hilden, Germany). Targeted genes were captured using Agilent (Santa Clara, CA) SureSelect probes or Integrated DNA Technologies (Coral, IL) xGen Lockdown probes at positions where SureSelect yield was inadequate. Next-generation sequencing10 was performed on the Illumina (San Diego, CA) MiSeq or HiSeq 2500 to at least 450× average coverage of 2 × 150 reads, with a minimum of 50× required at every targeted position. Stringent process controls were used to minimize read-depth variability, and up to eight anonymous blood samples were used as control specimens in each run to measure remaining coverage variability.11
Germline variant calling and assessment
Reads were aligned to the reference human genome sequence GRCh3712 using Novoalign (Novocraft Technologies, Selangor, Malaysia). Sequence variants were then analyzed for indels and single-nucleotide variants (SNVs) using the Genome Analysis Toolkit (GATK).13 Copy-number variants were called using read-depth analysis with CNVitae.11,14 Split-read analysis15 was performed as described previously16 to detect genomic structural variants. Candidate variants were classified as pathogenic (P) or likely pathogenic (LP) if they involved large genomic events or conferred a truncating, initiation codon or splice donor/acceptor effect; if functional data showed an impact on protein function; or if pathogenicity was otherwise reported in published literature.17 P and LP variants were orthogonally confirmed using Sanger sequencing or multiplex ligation-dependent probe amplification (MLPA)18 in accordance with Invitae standard operating practices.11 Confirmed variants were then interrogated using refined American College of Medical Genetics and Genomics criteria (Sherloc).19 In this study, “pathogenic” variants were defined as those classified as P or LP. Variants of unknown significance results were excluded from this study. For each of the examined genes, the frequency of pathogenic germline variants relative to the number of patients sequenced was calculated (Table S1.3, Fig. S1).
Selection of DRG pathways
Among the examined genes, genes with more than 100 total requisitions were analyzed (n = 83 genes, Tables S1.3, S1.4). DRGs (n = 22) were defined in this study by their presence in one of the following pathways as previously described by our group:20,21 homologous recombination, Fanconi, nucleotide excision repair, mismatch repair, and other (Table S1.4).
Enrichment analysis
For each of the examined DRGs, we calculated the frequency of pathogenic germline variants relative to the number of patients in which the gene was tested. We then performed enrichment analysis and compared the frequency of pathogenic germline variants in UC patients with the frequency of these gene variants in a population of cancer-free controls derived from ExAC.9 Since ancestry in our cohort was physician-reported, we followed special considerations when selecting the comparator control group from ExAC. For each gene, we selected the corresponding population in ExAC with the highest frequency of pathogenic germline variants in that gene. As an example, the East Asian ExAC population has the highest reported frequency of MSH2 germline variants among all ancestries (0.23%). As such, the East Asian ExAC population was used as the control group when comparing the prevalence of pathogenic MSH2 variants with our UC cohort.
For the enrichment analysis, we included all pathogenic variant calls from our cohort except for genomic alterations not included in ExAC data release that was used for the analysis (deletions [n = 15] or duplications [n = 1] spanning entire exons) or intronic regions without adequate coverage (n = 1). Of note, loss-of-function (LOF) variants in CHEK2 and low penetrance CHEK2 p.Ile157Thr variants were analyzed separately as they exhibit distinctive functional and clinical features.
Clinically actionable genes
Actionable genes were defined as established cancer predisposition genes that confer a higher risk for any cancer phenotype and for which enhanced screening and family genetic testing are recommended by the National Comprehensive Cancer Network (NCCN). APC, ATM, BRCA1, BRCA2, BRIP1, CHEK2, MLH1, MSH2, MSH6, MUTYH, PALB2, PMS2, RAD51C, RAD51D, and TP53 met these criteria.22,23
Statistical analysis
Two-sided Fisher's exact tests were used to calculate the odds ratios (OR), 95% confidence intervals (CI), and P values of all enrichment analyses. We applied Bonferroni correction for the number of independent tests conducted and with a significant p value cutoff of 0.05. Associations between pathogenic germline variants and gender, or site of UC were evaluated with the use of two-sided Fisher’s exact tests. The Mann–Whitney test was used to analyze associations with the age at diagnosis.
RESULTS
Patient characteristics
Data on 1038 patients with urothelial carcinoma (UC) of the bladder (923, 89%) or upper tract (67, 6%) were analyzed (Tables 1, S1.1). The site of UC was unknown for 48 (5%) patients. The mean age at testing was 58 years (range 6–89 years). Most patients were white non-Hispanics (787/1038, 76%). Multiple primary tumors were common: 672 (65%) patients had a personal history of another malignancy, excluding nonmelanoma skin cancers, with breast (n = 192, 18%), prostate (n = 173, 17%), and colon (n = 128, 12%) being the most common. In addition, 113 patients (11%) reported one or more first- or second-degree relatives with UC (Table 1).
Germline genomic landscape of urothelial carcinoma
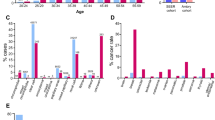
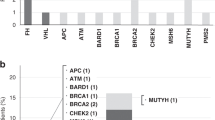
For the genes analyzed (mean = 45, median = 42, range = 1–130), 203 pathogenic variants were reported. The cumulative frequency of patients with pathogenic germline variants in all examined genes was 24%. Overall, the highest frequency of pathogenic germline variants was in MSH2 (34/969, 3.5%, 95% CI = 2.5–4.9%), BRCA1 (20/867, 2.3%, 95% CI = 1.5–3.5%), BRCA2 (18/867, 2.1%, 95% CI = 1.3–3.3%), heterozygous MUTYH (15/754, 2.0%, 95% CI = 1.2–3.3%), and ATM (13/827, 1.6%, 95% CI = 0.9–2.7%) as shown in Tables 2 and S1.3. Loss-of-function and low penetrance CHEK2 variants were each identified in 1.4% (12/862, 95% CI = 0.8–2.4%) of patients. There were diverse variant types observed by gene (Fig. 1, Table S1.2). Of note, fumarate hydratase (FH) pathogenic germline variants were found in 1.3% (5/390, 95% CI = 0.5–3.0%) while germline variants in the melanocyte inducing transcription factor (MITF) were detected in 1.2% (4/339, 95% CI = 0.5–3.0%, Table S1.3). Among the five FH germline carriers, none had a diagnosis of renal cell carcinoma. Of the four MITF p.E318K carriers, only one patient had a personal history of an MITF-associated malignancy (melanoma) (Table S1.1).

Locations of variants and domains in proteins encoded by 11 DRGs are shown by lollipop structures, with the variant class indicated by different colors. Protein domains are also shown in different colors. For each gene, the x-axis reflects the number of amino acid residues, and the y-axis shows the total number of pathogenic variant counts.
Among the 366 UC patients who did not have a personal history of a second malignancy, the frequency of pathogenic germline variants in all the genes analyzed was 24.4%. In this subset, germline genetic variants were most frequent in MSH2 (13/339, 3.8%, 95% CI = 2.3–6.5%), BRCA1 (10/281, 3.6%, 95% CI = 1.9–6.4%), and heterozygous MUTYH (7/246, 2.8%, 95% CI = 1.4–5.8%).
Pathogenic germline variants in DRGs
Among the 1038 patients, 20% harbored pathogenic germline variants in one of the DRGs. The frequency of germline variants in DRGs was 19.3% among patients with UC only (n = 366). Pathogenic variants in the homologous recombination (HR) pathway were the most prevalent, occurring at a cumulative frequency of 11.1% (Table S1.4, Fig. S2). Among the HR pathway genes, a total of 20 pathogenic variants were identified in BRCA1 and another 18 pathogenic variants were found in BRCA2. Of those, 18 of 20 (90%, CI = 70–98%) and 16 of 18 (89%, CI = 67–98%) carried LOF variants (Fig. 1, Tables 2, S1.2), respectively. Additionally, our analysis identified 13 pathogenic variants in ATM. Of those, 12 (92%, CI = 67–100%) caused LOF either by a protein truncating effect (n = 11) or by amino acid substitution in the functionally critical FAT domain (n = 11) (Figs. 1, 2, Table S1.2).

(a) Enrichment of pathogenic germline damage repair gene (DRG) variants in individuals with urothelial carcinoma. Fisher’s exact test was used to calculate the odds ratios (ORs) and 95% confidence intervals (CIs). A two-sided binomial test was used to compute the p values. (b) Applying a false discovery rate of less than 0.05 (genes above the red dotted line), MSH2, ATM, MLH1, and BRCA2 showed significant enrichment of pathogenic germline variants in the urothelial carcinoma cohort compared with the corresponding cancer-free populations showing the highest frequency of variants for each gene.
The mismatch repair (MMR) pathway had the next highest frequency of pathogenic variants. MMR genes, including MLH1, MSH2, MSH6, and PMS2, are associated with an increased risk of urothelial carcinoma.7 Pathogenic germline variants in these genes were detected in 5.9% of patients and represented a quarter of the total germline variants detected in DRGs. The classes of germline variants and their loci relative to the domains for all four Lynch syndrome genes were evaluated (Fig. 1). All MSH2 pathogenic variants (n = 34) were predicted to cause LOF with 11 (32%, CI = 19–49%) MSH2 pathogenic calls involving exonic deletions. Six of 10 (60%, CI = 31–83%) pathogenic variants in MLH1 led to protein truncation. One variant p.Gly67Arg affected the histidine kinase-, DNA gyrase B, and HSP90-like ATPase (HATPase_c_3). Among the eight germline variants in MSH6, six (75%, CI = 41–96%) were truncating. Both pathogenic germline variants in PMS2 were missense and involved either the HATPase- or MutL-C terminal domains (Fig. 1).
Enrichment of germline DNA damage repair variants in urothelial carcinoma
Based on data from prior studies suggesting higher frequency of germline defects in the MMR pathway in UC, we performed enrichment analysis on an expanded set of 17 DRGs (see “Materials and Methods”). Gene coverage, various population groups, and their cohort sizes are shown in Supplementary Tables S1.5 and S1.6. Using a p value of less than 0.05 for significant associations (correcting for the number of conducted tests), we showed a significant pathogenic variant enrichment of ATM, BRCA2, MLH1, and MSH2 in our cohort relative to cancer-free cohorts (Fig. 2a, b, and Table 2). Systematic underestimation of the observed versus expected p value distribution was noted for the other genes analyzed, suggesting that a stringent analytic approach was followed (Fig. S3).
Germline pathogenic variants in significantly enriched known UC risk genes
Our enrichment analysis findings showed that patients with germline alterations in MLH1 and MSH2 were roughly 15 times more likely to develop UC compared with cancer-free controls (OR = 15.9, 95% CI = 4.4–67.7, adjusted p < 0.0001 and OR = 15.4, 95% CI = 7.1–32.7, adjusted p < 0.0001, respectively). Of note, 7 of 10 (70%, CI = 40–89%) pathogenic variants in MLH1 led to LOF.
Germline pathogenic variants in additional significantly enriched DRGs
In addition to MLH1 and MSH2, our analysis also identified significant enrichment in two other DRGs, ATM and BRCA2. Compared with cancer-free individuals, patients with UC were roughly six times more likely to carry pathogenic variants in BRCA2 (OR = 5.7, 95% CI = 3.2–9.6, p < 0.0001). Our enrichment analysis also demonstrated that germline pathogenic variants in ATM were approximately four times more frequent in the UC patients compared with the ExAC cohort (OR = 3.8, 95% CI = 1.8–8.3, adjusted p = 0.02).
Actionable germline variants in urothelial carcinoma
The prevalence of actionable pathogenic variants in UC patients was 18.6%. Lynch syndrome gene variants represented 5.9% of the total 18.6% (Table 3). Highly penetrant cancer risk variants were as follows: BRCA1 or BRCA2 (38/867, 4.4%, 95% CI = 3.2–5.9%), MSH2 (34/969, 3.5%, 95% CI = 2.5–4.9%), MLH1 (10/957, 1%, 95% CI = 0.6–1.9%), and TP53 (2/929, 0.2%, 95% CI = 0.04–0.8%). The most common moderately penetrant actionable pathogenic variants were CHEK2 (12/862, 1.4%, 95% CI = 0.8–2.4%), MSH6 (8/959, 0.8%, 95% CI = 0.4–1.6%), and PMS2 (5/956, 0.5%, 95% CI = 0.2–1.2%). Low penetrance variants were most frequent as follows: CHEK2, p.I157T variant (12/862, 1.4%, 95% CI = 0.8–2.4%) and heterozygous MUTYH (15/754, 2%, 95% CI = 1.2–3.3%, Tables 3 and S1.7).
Association between germline variant carrier status and clinical factors
Four genes (MSH2, BRCA1, BRCA2, and MUTYH) met criteria for this analysis as they had over 100 requisitions and pathogenic variants were present in ≥2% of UC patients. There was no enrichment of MSH2 carriers across age, gender, or family history of any cancer. Patients with upper tract urothelial carcinoma (UTUC) were more likely to have MSH2 pathogenic variants compared with those with bladder tumors only (9/67, 13% versus 22/856, 3%; OR = 5.9; 95% CI = 2.6–13.4; p < 0.001). BRCA1, BRCA2, and MUTYH pathogenic variants did not correlate with age at diagnosis, gender, family history of any cancer, or site of UC. Furthermore, patients with a history of another malignancy in addition to UC were not enriched for any of the four genes when compared with those with a sole diagnosis of UC. Of the female patients with a personal history of breast cancer, 4 of 176 (2.3%) had a BRCA2 germline variant. In comparison, 4 of 266 (1.5%) patients who did not have a diagnosis of breast cancer harbored BRCA2 pathogenic germline variants. Similarly, female patients with a personal history of breast cancer were not significantly more likely to harbor BRCA1 pathogenic variants compared with females who did not have a diagnosis of breast cancer (OR = 1.3, 95% CI = 0.4–4.2, p = 0.76).
DISCUSSION
Several published studies have reported on somatic drivers of urothelial carcinoma.24,25 Others have correlated such drivers with therapeutic outcomes and elucidated novel potential biomarkers. For example, DRGs have been shown to enhance sensitivity to platinum-based regimens and immune checkpoint therapy in both neoadjuvant26,27 and metastatic settings.28,29 However, so far, the prevalence and significance of germline drivers of UC have not been fully characterized. Herein, our analysis of 1038 patients with cancer of the bladder or upper tract revealed a prevalence of germline variants in 24% of patients with the majority (20%) harboring variants in DRGs. This finding is consistent with prior studies though prior studies are limited by small numbers or enriched by patients with advanced disease. One study of East Asian bladder cancer patients (n = 98) found pathogenic and likely pathogenic variants in DRGs occurred in 10.2%.5 Another report of 101 UC patients sequenced using a 76-gene panel (MSK-IMPACT) revealed pathogenic or likely pathogenic germline variants in 22% of patients. Of the 101 patients, 27% had metastatic disease.30
In this cohort, we reported significantly higher rates of germline pathogenic variants in ATM, BRCA2, MLH1, and MSH2 relative to cancer-free control individuals. MLH1 and MSH2 have been correlated with heritable risks for UC.7 The power of the size of our cohort enabled the discovery of other significantly altered genes. Beyond MLH1 and MSH2, our analysis identified significant enrichment in two other genes (ATM and BRCA2) that have not been previously associated with a higher risk of UC. ATM encodes a protein kinase and is an essential component of the HR pathway. In response to double-strand breaks in DNA, ATM phosphorylates downstream cellular processes that ultimately regulate DNA-end resection.31 Prior studies have focused on ATM’s pivotal role in cancer development and associated heterozygous carriers of ATM variants with increased risks of breast cancer, colorectal cancer,20 and potentially pancreatic cancer.32 BRCA2 is another HR pathway master regulator that encodes a protein that mediates the recruitment of the recombinase RAD51 to DNA double-stranded breaks.33 Although prior studies clearly linked pathogenic ATM and BRCA2 variants to the development of several cancers, none have associated such variants with an increased risk of UC. However, adequately controlled enrichment studies (where genomic data of cases and controls are jointly analyzed and robustly stratified for ancestry) are still needed to validate such findings and to delineate the potential gene-specific UC risk.
Germline variants in DRGs were found in 20% of patients, among which HR pathway genes contributed to almost half of the DRG variants. These results are strikingly consistent with prior work that showed germline alterations in DRGs were present in 19% of patients with UC. The same group also showed that variants in HR and MMR pathway genes occur in 11/101 (11%) and 8/101 (8%) patients, respectively.30 Similarly, an analysis of 1040 patients with several cancers showed that among 16 patients with bladder cancer, 2 (13%) harbored pathogenic germline variants in the HR pathway.34 In a separate study5 of 98 East Asian patients with UC with all stages of disease who were referred for multi-DRG germline testing (n = 54 DRGs), 10.2% harbored pathogenic DRG variants, 1% had a pathogenic variant in an MMR gene, while 4 (4%) had variants in HR pathway genes. The differences in observed variant frequencies from our study may be due to variant calling processes or ancestry.
Recent studies have linked germline variants in canonical HR genes with substantial responses to poly (ADP ribose) polymerase (PARP) inhibitors,35,36 which augments DNA damage and leads to tumor cell death.37 Ongoing trials (NCT02286687, NCT02286687) are also studying the association between HR genes and other DRGs with clinical outcomes in cancer patients treated with PARP inhibitors. Collectively, 5% of the UC patients in our study carried a variant in BRCA1, BRCA2, or PALB2, which are core components of the HR pathway. Additionally, our study found 5.9% of patients with germline MMR variants. This is especially pertinent in the setting of immune checkpoint inhibitors, which are currently approved for all solid cancers with evidence of MMR deficiency.
Taken together, a total of 11% of UC patients in our cohort had germline variants that may be informative for therapeutic decision making. In addition to the therapeutic utility, variants in these genes significantly increase a patient’s lifetime risk of developing other malignancies, some of which have readily available and effective screening options. Moreover, identifying pathogenic variant carriers may initiate cascade genetic testing among at-risk relatives for risk stratification and tailored health management.
We also found pathogenic germline variants in FH and MITF in 1.3% and 1.2% respectively. Pathogenic germline variants in FH cause hereditary leiomyomatosis and renal cell carcinoma (HLRCC), a syndrome associated with papillary RCC and uterine and cutaneous leiomyomas. There have been two prior reports of FH pathogenic germline variants in UC patients.30,38 The MITF p.E318K variant has been associated with both melanoma and renal cell carcinoma.39 Future studies focused on screening genetically enriched patients may be a powerful strategy to determine whether this variant or the FH variants confer an increased risk of urothelial carcinoma.
The heritability of bladder cancer is estimated to be 30% based on twin studies.2 In our analysis, germline pathogenic variants in Lynch syndrome genes explained the higher risk of UC in only 6% of highly selected UC patients, leaving much of the missing heritability unexplained. Some may be explained by variants in other genes, such as DRGs involved in the HR pathway. However, future case–control studies of selected and unselected cohorts are required to explore novel UC risk genes.
Our study has several limitations. First, our patients were selected for genetic testing based on a suspicion of heritable pathogenic variants: history of ≥1 other nonurothelial cancers, young age, or family history of cancer or cancer predisposition syndrome. Hence, our findings may not be generalizable to unselected UC cohorts. Second, several ethnic populations were underrepresented in this cohort including Hispanics, Asians, and African Americans, which is typical of genetic studies and has been attributed to pervasive disparities in this field.40 Third, family history information was not directly derived from medical records, but rather was gathered from test requisitions, a method that can be prone to errors of omission. Fourth, data on other drivers of UC including smoking history and occupational exposures were not obtained. The stage of disease was not available, which precluded analysis germline variants by stage. Fifth, the raw genomic data were not available for analysis, which limited the ability to perform ancestry inference or joint genetic analysis and may have introduced unintended bias to the enrichment analysis. To mitigate this, we chose the ExAC cohort with maximum gene alterations as a control group. However, differences in sequencing depth of coverage and genomic interval capturing cannot be fully controlled in such analyses. Despite this being the largest study to date of UC patients completing germline genetic testing, there remain small numbers of pathogenic variant carriers per gene, making genotype–phenotype associations difficult. Paired tumor sequencing was not available and thus germline drivers of tumorigenesis were difficult to determine. Finally, given the variability in genes tested per patient, we limited the analysis to a subset of high-impact genes.
Conclusion
In this large study evaluating 1038 UC patients, 24% of patients carried pathogenic cancer predisposition variants with 20% harboring variants in DNA damage repair genes. In addition, this study prioritized DRGs with potential pathogenic variant enrichment that could be explored as UC risk genes in prospective case–control studies. While 11% of tested UC patients were found to have germline variants for which targeted therapeutics may be considered, actionable germline variants that have gene-specific NCCN cancer management recommendations were identified in 19% of UC patients. Overall, our data suggest a potential clinical utility of germline analysis of selected UC patients. Furthermore, studies are required to determine the effects of precision therapeutics targeting germline variants on outcomes in individuals with UC.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34.
Mucci LA, Hjelmborg JB, Harris JR, et al. Familial risk and heritability of cancer among twins in Nordic countries. JAMA. 2016;315:68–76.
Martin C, Leiser CL, O'Neil B, et al. Familial cancer clustering in urothelial cancer: a population-based case-control study. J Natl Cancer Inst. 2018;110:527–533.
Huang KL, Mashl RJ, Wu Y, et al. Pathogenic germline variants in 10,389 adult cancers. Cell. 2018;173:355–370, e314.
Na R, Wu Y, Jiang G, et al. Germline mutations in DNA repair genes are associated with bladder cancer risk and unfavourable prognosis. BJU Int. 2018;122:808–813.
Skeldon SC, Semotiuk K, Aronson M, et al. Patients with Lynch syndrome mismatch repair gene mutations are at higher risk for not only upper tract urothelial cancer but also bladder cancer. Eur Urol. 2013;63:379–385.
Engel C, Loeffler M, Steinke V, et al. Risks of less common cancers in proven mutation carriers with lynch syndrome. J Clin Oncol. 2012;30:4409–4415.
van der Post RS, Kiemeney LA, Ligtenberg MJ, et al. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J Med Genet. 2010;47:464–470.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291.
Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59.
Lincoln SE, Kobayashi Y, Anderson MJ, et al. A systematic comparison of traditional and multigene panel testing for hereditary breast and ovarian cancer genes in more than 1000 patients. J Mol Diagn. 2015;17:533–544.
Church DM, Schneider VA, Graves T, et al. Modernizing reference genome assemblies. PLoS Biol. 2011;9:e1001091.
McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303.
Truty R, Paul J, Kennemer M, et al. Prevalence and properties of intragenic copy-number variation in Mendelian disease genes. Genet Med. 2019;21:114–123.
Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12:363–376.
Nord AS, Lee M, King MC, Walsh T. Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genomics. 2011;12:184.
Kurian AW, Hare EE, Mills MA, et al. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol. 2014;32:2001–2009.
Hogervorst FB, Nederlof PM, Gille JJ, et al. Large genomic deletions and duplications in the BRCA1 gene identified by a novel quantitative method. Cancer Res. 2003;63:1449–1453.
Nykamp K, Anderson M, Powers M, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017;19:1105–1117.
AlDubayan SH, Giannakis M, Moore ND, et al. Inherited DNA-repair defects in colorectal cancer. Am J Hum Genet. 2018;102:401–414.
AlDubayan SH, Pyle LC, Gamulin M. et al. Association of inherited pathogenic variants in checkpoint kinase 2 (CHEK2) with susceptibility to testicular germ cell tumors. JAMA Oncol. 2019;5:514–522.
Daly MB, Pilarski R, Berry M, et al. NCCN guidelines insights: genetic/familial high-risk assessment: breast and ovarian, version 2.2017. J Natl Compr Canc Netw. 2017;15:9–20.
Gupta S, Provenzale D, Regenbogen SE, et al. NCCN guidelines insights: genetic/familial high-risk assessment: colorectal, version 3.2017. J Natl Compr Canc Netw. 2017;15:1465–1475.
Robertson AG, Kim J, Al-Ahmadie H, et al. Comprehensive molecular characterization of muscle-invasive bladder. Cancer. Cell. 2017;171:540–556, e525.
Moss TJ, Qi Y, Xi L, et al. Comprehensive genomic characterization of upper tract urothelial carcinoma. Eur Urol. 2017;72:641–649.
Van Allen EM, Mouw KW, Kim P, et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov. 2014;4:1140–1153.
Necchi A, Anichini A, Raggi D, et al. Pembrolizumab as neoadjuvant therapy before radical cystectomy in patients with muscle-invasive urothelial bladder carcinoma (PURE-01): an open-label, single-arm, phase II study. J Clin Oncol. 2018 Oct 20; https://doi.org/10.1200/JCO.18.01148 [Epub ahead of print].
Teo MY, Bambury RM, Zabor EC, et al. DNA damage response and repair gene alterations are associated with improved survival in patients with platinum-treated advanced urothelial carcinoma. Clin Cancer Res. 2017;23:3610–3618.
Teo MY, Seier K, Ostrovnaya I, et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD-1/PD-L1 blockade in advanced urothelial cancers. J Clin Oncol. 2018;36:1685–1694.
Carlo MI, Zhang L, Mandelker D, et al. Cancer predisposing germline mutations in patients (pts) with urothelial cancer (UC) of the renal pelvis (R-P), ureter (U) and bladder (B). J Clin Oncol. 2017;35(15_suppl):4510.
Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell. 2017;66:801–817.
Thompson D, Duedal S, Kirner J, et al. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005;97:813–822.
Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2011;12:68–78.
Mandelker D, Zhang L, Kemel Y, et al. Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA vs guideline-based germline testing. JAMA. 2017;318:825–835.
Litton JK, Rugo HS, Ettl J, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379:753–763.
Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–250.
O'Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015;60:547–560.
Lehtonen HJ, Kiuru M, Ylisaukko-Oja SK, et al. Increased risk of cancer in patients with fumarate hydratase germline mutation. J Med Genet. 2006;43:523–526.
Bertolotto C, Lesueur F, Giuliano S, et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature. 2011;480:94–98.
Armstrong K, Micco E, Carney A, Stopfer J, Putt M. Racial differences in the use of BRCA1/2 testing among women with a family history of breast or ovarian cancer. JAMA. 2005;293:1729–1736.
Acknowledgements
We thank all individuals who participated in this study. This work was supported by an American Society of Clinical Oncology (ASCO) Conquer Cancer Foundation Career Development Award (CCF CDA) CDA#13167 (S.H.A.) and a Prostate Cancer Foundation Young Investigator Award (18YOUN02) (S.H.A.).
Disclosure
K.W.M. reports consulting fees from Pfizer, EMD Serono, and OncLive as well as research funding from Pfizer. D.J.K. reports serving as a consultant for Novartis. T.K.C. reports receiving institutional and personal funds from Analysis Group, AstraZeneca, Alexion, Bayer, Bristol-Myers Squibb/ER Squibb and sons LLC, Cerulean, Eisai, Foundation Medicine Inc., Exelixis, Ipsen, Tracon, Genentech, Roche, Roche Products Limited, F. Hoffmann-La Roche, GlaxoSmithKline, Lilly, Merck, Novartis, Peloton, Pfizer, Prometheus Labs, Corvus, Calithera, Sanofi/Aventis, Takeda; reported receiving honoraria from the Analysis Group, AstraZeneca, Alexion, Sanofi/Aventis, Bayer, Bristol-Myers Squibb/ER Squibb and sons LLC, Cerulean, Eisai, Foundation Medicine Inc., Exelixis, Genentech, Roche, Roche Products Limited, F. Hoffmann-La Roche, GlaxoSmithKline, Heron Therapeutics, Lilly, Merck, Novartis, Peloton, Pfizer, EMD Serono, Prometheus Labs, Corvus, Ipsen, UpToDate, NCCN, Analysis Group, NCCN, Michael J. Hennessy (MJH) Associates, Inc. (Healthcare Communications Company with several brands such as OncLive, PeerView, and PER), L-path, Kidney Cancer Journal, Clinical Care Options, Platform Q, Navinata Healthcare, Harborside Press, American Society of Medical Oncology, NEJM, Lancet Oncology; having a consulting or advisory role at Analysis Group, AstraZeneca, Alexion, Sanofi/Aventis, Bayer, Bristol-Myers Squibb/ER Squibb and sons LLC, Cerulean, Eisai, Foundation Medicine Inc., Exelixis, Genentech, Heron Therapeutics, Lilly, Roche, GlaxoSmithKline, Merck, Novartis, Peloton, Pfizer, EMD Serono, Prometheus Labs, Corvus, Ipsen, UpToDate, NCCN. No speaker's bureau. No leadership or employment in for-profit companies. Other present or past leadership roles: Director of GU Oncology Division at Dana-Farber and past President of Medical Staff at Dana-Farber), member of NCCN Kidney Panel and the GU Steering Committee, past chairman of the Kidney Cancer Association Medical and Scientific Steering Committee). Patents, royalties or other intellectual properties: International Patent Application number PCT/US2018/12209, entitled “PBRM1 Biomarkers Predictive of Anti-Immune Checkpoint Response,” filed 3 January 2018, claiming priority to US Provisional Patent Application number 62/445,094, filed 11 January 2017; International Patent Application number PCT/US2018/058430, entitled “Biomarkers of Clinical Response and Benefit to Immune Checkpoint Inhibitor Therapy,” filed 31 October 2018, claiming priority to US Provisional Patent Application number 62/581,175, filed 3 November 2017. Travel, accommodations, expenses, in relation to consulting, advisory roles, or honoraria. Medical writing and editorial assistance support may have been funded by communications companies funded by pharmaceutical companies (ClinicalThinking, Envision Pharma Group, Fishawack Group of Companies, Health Interactions, and Parexel, others). The institution (Dana-Farber Cancer Institute) may have received additional independent funding of drug companies or/and royalties potentially involved in research around the subject matter. Curriculum vitae provided upon request for scope of clinical practice and research. L.C.H. reports consulting fees from Genentech, Dendreon, Pfizer, Medivation/Astellas, Exelixis, Bayer, Kew Group, Corvus, Merck, Novartis, Michael J. Hennessy Associates (Healthcare Communications Company and several brands such as OncLive and PER), Jounce, and EMD Serono; Research funding from Bayer, Sotio, Bristol-Myers Squibb, Merck, Takeda, Dendreon/Valient, Jannsen, Medivation/Astellas, Genentech, Pfizer, Endocyte (Novartis), and support for research travel from Bayer and Genentech. N.M.C. reports support for research travel from Pfizer and Ipsen and consulting fees for BMS. E.V.A. reports serving as an advisor or consultant for Tango Therapeutics, Genome Medical, Invitae, Illumina, Foresite Capital, and Dynamo. He receives research support from Novartis, BMS; has equity in Tango Therapeutics, Genome Medical, Syapse, and Microsoft; receives travel reimbursement from Roche/Genentech; and holds institutional patents on ERCC2 variants and chemotherapy response, chromatin variants and immunotherapy response, and methods for clinical interpretation. S.Y., E.D.E., and T.C. are employees of Invitae Corporation, a testing laboratory that furnished the diagnostic test results used in this study. S.Y. reports other support from Invitae during the conduct of the study. E.D.E. reports being a shareholder of Invitae. J.E.G.: AstraZeneca, Helix Genetics, Ambry Genetics, Invitae Genetics, Myriad Genetics, Konica-Minolta, Novartis Pharmaceuticals, H.Q.R.: received honoraria from Ambry Genetics. G.S.: consultant for BMS, Exelixis, Bayer, Sanofi, Pfizer, Novartis, Eisai, Janssen, Amgen, AstraZeneca, Merck, Genentech, EMD Serono, Astellas/Agensys; research support to institution from AstraZeneca, Bayer, Amgen, Boehringer-Ingelheim, Janssen, Merck, Sanofi, Pfizer; author for UpToDate; Steering Committee for AstraZeneca, BMS, Bavarian Nordic, Astellas; speaker for OncLive; Research to Practice; Physician Education Resource (PER). The other authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nassar, A.H., Abou Alaiwi, S., AlDubayan, S.H. et al. Prevalence of pathogenic germline cancer risk variants in high-risk urothelial carcinoma. Genet Med 22, 709–718 (2020). https://doi.org/10.1038/s41436-019-0720-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0720-x
Keywords
This article is cited by
-
A chromosome-scale fishing cat reference genome for the evaluation of potential germline risk variants
Scientific Reports (2024)
-
Global trends in the epidemiology of bladder cancer: challenges for public health and clinical practice
Nature Reviews Clinical Oncology (2023)
-
Cross-species oncogenomics offers insight into human muscle-invasive bladder cancer
Genome Biology (2023)
-
The germline mutational landscape of genitourinary cancers and its indication for prognosis and risk
BMC Urology (2022)
-
Germline BRCA1 and BRCA2 mutations and the risk of bladder or kidney cancer in Poland
Hereditary Cancer in Clinical Practice (2022)

{kind=link}