
Abstract
Purpose
The ability of a single technology, next-generation sequencing, to provide both sequence and copy number variant (CNV) results has driven the merger of clinical cytogenetics and molecular genetics. Consequently, the distinction between the definition of a sequence variant and a CNV is blurry. As the 2015 American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) standards and guidelines for interpretation of sequence variants address CNV classification only sparingly, this study focused on adapting ACMG/AMP criteria for single-gene CNV interpretation.
Methods
CNV-specific modifications of the 2015 ACMG/AMP criteria were developed and their utility was independently tested by three diagnostic laboratories. Each laboratory team interpreted the same 12 single-gene CNVs using three systems: (1) without ACMG/AMP guidance, (2) with ACMG/AMP criteria, and (3) with new modifications. A replication study of 12 different CNVs validated the modified criteria.
Results
The adapted criteria system presented here showed improved concordance and usability for single-gene CNVs compared with using the ACMG/AMP interpretation guidelines focused on sequence variants.
Conclusion
These single-gene CNV criteria modifications could be used as a supplement to the ACMG/AMP guidelines for sequence variants, allowing for a streamlined workflow and a step toward a uniform classification system for both sequence and copy number alterations.
Similar content being viewed by others


INTRODUCTION
Today the power of a single technology, next-generation sequencing (NGS), to provide both sequence and copy number variant (CNV) results has become a driving force for the merger of clinical cytogenetics and molecular genetics. Consequently, the distinction between sequence and copy number variants is blurred and exon-level deletion/duplication events can be considered to fall into either category. Since both types of variants can be identified and reported by a single test, maintaining a distinction between these types is difficult and needless. Moreover, CNVs are a significant cause of genetic disorders, representing 5–9% of pathogenic/likely pathogenic variants in multigene panel testing for various disorders.1,2 The majority of these CNVs are multiexon deletions, followed by single-exon deletions and multiexon duplications.2 Reflective of this merger between sequence and copy number variant detection, the American Board of Medical Genetics and Genomics (ABMGG) has merged cytogenetic and molecular genetic subspecialties into a combined training program.3 Movement toward unified interpretation of all types of genetic alterations is critical for laboratory efficiency and consistency, as well as for the generation of a clear, comprehensive test report.
The standards and guidelines for the interpretation of sequence variants, created jointly by the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) in 2015 (ACMG/AMP guidelines), address the classification of sequence variants based on a semiquantitative criteria system; however, single-gene CNVs were addressed sparingly (e.g., PVS1) and multigene CNVs identified by chromosomal microarray were not included.4 In this study we aimed to extend the ACMG/AMP guidelines to CNVs involving single genes. Many criteria were directly applicable with only minor customizations (e.g., criteria considering parentage or segregation). Nevertheless, there are interpretation challenges unique to CNVs. For example, CNV interpretation utilizes specialized resources, e.g., DECIPHER and the Database of Genomic Variants (DGV), with different limitations compared with sequence variant databases.5,6 It can be more difficult to compare a proband’s CNV with published CNVs in the same region due to breakpoint uncertainty, often resulting from the use of diverse technologies. Copy number gain interpretation is further complicated when location and orientation are unknown. Single-gene CNV criteria modifications presented here were tailored to address these challenges and further honed by testing on hundreds of different CNVs across >100 genes. In addition, we compared classifications assigned to CNVs across three independent laboratories without 2015 ACMG/AMP guidelines versus with ACMG/AMP guidelines versus with single-gene CNV modifications of these guidelines. Unification of sequence and single-gene copy number variant interpretation could be expanded to multigenic CNVs in the future.
MATERIALS AND METHODS
To assess which of the ACMG/AMP guidelines criteria would need to be adapted concerning single-gene CNV mechanisms, we examined 454 consecutive CNVs found by targeted array comparative genomic hybridization (aCGH) in multigene sequencing plus deletion/duplication testing at one laboratory. These CNVs were stratified by exon involvement and CNV type. Variants observed to extend into neighboring genes due to flanking backbone probe deviation were excluded. Arrays included coverage of most coding exons of panel genes and, for some genes, intronic regions. Several CNV interpretation experts at this laboratory used an iterative process to adapt the ACMG/AMP guidelines classification system to fit this broad spectrum of CNVs.
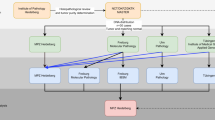
Twenty-four variants were provided for an intra- and interlaboratory study to evaluate the utility and ease of use of adapted criteria; these variants represent diverse disease mechanisms, inheritance patterns, and phenotypes and reflect the variant types identified among 454 consecutive CNVs (study design summarized in Fig. 1). Interpretation of these CNVs was independently performed by at least five teams with single-gene CNV vetting experience, each from a clinical diagnostic laboratory (GeneDx, EGL Genetics [formerly Emory Genetics Laboratory], or University of Chicago Genetic Services Laboratory). Participants included a certified genetic counselor, variant interpretation scientists (n = 3), and laboratory directors (ABMGG-certified in molecular genetics [n = 4] or molecular and cytogenetics [n = 2]). Years in practice ranged from 1.5 to 15 (median 5.5). Study participants were provided the description of each variant according to the International System of Human Cytogenomic Nomenclature (ISCN) 20167 and the clinical information in Tables 1 and S1. Nonpublished clinical or familial data provided in this study were simulated to evaluate utility of criteria. Teams of one to two participants were instructed to interpret each variant in the context of an affected patient undergoing a clinical test including the involved gene. Teams completed three iterative rounds of interpretation of the same 12 variants (Table 1). Round 1 required each team to use their lab-internal (non-ACMG/AMP) classification systems. Round 2 utilized 2015 ACMG/AMP guidelines alone.4 Round 3 was performed using ACMG/AMP guidelines criteria adjusted for single-gene CNVs (Table S2). Classification concordance was then evaluated. Pathogenic (P)/likely pathogenic (LP) versus uncertain significance (VUS)/likely benign (LB)/benign (B) differences were considered medically significant.8 In addition, participants completed a survey regarding their experiences across the different systems. Finally, validation of the modified criteria was carried out by repeating rounds 1 and 3 using a different set of 12 variants (Table S1, replication study).

Study design for testing classification of single-gene copy number variants (CNVs) across multiple systems and laboratories. Teams completed three iterative rounds of interpretation of the same 12 variants. Round 1 required each laboratory to use their historic, non–American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines internal or laboratory-developed classification systems. Round 2 utilized the 2015 ACMG/AMP guidelines publication alone.4 Round 3 was performed using ACMG/AMP guidelines modifications for single-gene CNVs. Rounds 1 and 3 were then repeated on a new set of 12 variants (replication study).
RESULTS
The spectrum of single-gene CNVs observed in clinical practice was compiled to assess which of the ACMG/AMP guidelines criteria would need to be adapted concerning single-gene CNV mechanisms. Figure 2 illustrates the frequency and types of CNVs observed across 454 consecutive single-gene CNVs from cases submitted for a variety of multigene tests, representing 164 different genes and 318 unique CNVs. Approximately 43% of the 454 CNVs were gains (n = 193) and ~57% were losses (n = 261). Among unique CNVs, ~32% were gains and 68% were losses. Of the total observed CNVs, 22% appeared to involve a single exon (n = 100), 60% were multiexonic (n = 271), and 18% did not involve primary transcript exons (n = 83). The most commonly observed variant types were intragenic multiexon losses (~20%, n = 92), followed by intronic gains (~13%, n = 61), intragenic single exon losses (~12%, n = 55), and gains/losses involving multiple 3’ exons (each ~9%, n = 43/n = 39, respectively). The genes included in this analysis are associated with diverse disease mechanisms, inheritance patterns, and phenotypes. Most deletions affecting exonic sequence were classified as P/LP while most duplications affecting exonic sequence were classified as VUS (Fig. S1). All CNV types listed in Fig. 2 and their unique challenges were considered in the formulation of the CNV-adjusted ACMG/AMP guidelines criteria. The final modifications are summarized in Table S2 and described in the following sections. These modified criteria were most commonly applied, indicating importance: PVS1, PM2, PM4_Strong, BA1, and BS1 (Table S3).

Frequency and types of single-gene copy number variants (CNVs) observed during multigene panel testing. Data represent 454 consecutive single-gene CNVs observed in an approximately six-month time period by a wide variety of multigene panel testing. The Y-axis is the number of observations and the X-axis is the type of CNV. Copy number gains labeled “intragenic” are most often presumed intragenic as location/orientation of the additional material was unknown in the majority of cases. Copy number variants in the “other” category include those involving promoter regions, intronic sequence, or alternative transcripts.
PVS1
The PVS1 criterion, “Null variant (nonsense, …, single or multiexon deletion) in a gene where LOF is a known mechanism of disease,”4 was adapted relative to CNV type and size. This designation of “very strong” is based on the well-established model that variants resulting in premature stop codons typically result in nonsense-mediated decay (NMD) of the messenger RNA (mRNA) and produce no/minimal amounts of truncated protein.9,10 To safeguard against an overclassification of variants that might unexpectedly produce a full or partially functional protein (e.g., escape NMD), use of PVS1 alone will not result in a P classification. However, when a deletion affects all known coding exons of a disease gene where loss of function (LOF) is the definitive mechanism of disease, this safeguard results in underclassification in the absence of other moderate or supporting evidence. Thus, for this scenario, PVS1 could be strength-modified to a pathogenic criterion that, when used alone, results in a P classification (PVS1_Stand-alone). A proposal on adjusting the strength of PVS1 and PM4 is further illustrated in Fig. 3.

Modifications to PVS1 and PM4 for deletions and duplications affecting genes with known loss of function (LOF) disease mechanisms. In the absence of functional studies, there is no applicable pathogenic criterion for a deletion/duplication involving a gene known to have a disease mechanism other than LOF. PVS1_Moderate may be used for a CNV resulting in LOF of a gene with unknown disease mechanism. No pathogenic criterion is applicable for duplications involving the 5’ and/or 3’ end of the gene without additional studies characterizing actual effect. For duplications with known breakpoints proving that the event is not in tandem (e.g., from sequencing data), applicable criteria depend on exact molecular nature.
Other multiexon deletions often lead to a null allele, even when not involving all coding exons. It is prudent to consider the impact on the reading frame to predict whether or not the mRNA would be subject to NMD; however, for larger multiexonic deletions, this calculation may become less relevant and may be inaccurate if there is uncertainty regarding involvement of entire exons and/or neighboring exons. Regardless of frame, if the deletion involves a region critical to protein function (i.e., would generate a functional null allele) in a gene with a LOF disease mechanism, then PVS1 usage is also applicable. Additionally, multiexon deletions that remove a substantial fraction of the coding region of the biologically relevant transcript are likely to produce a functionally disrupted protein at a minimum, supporting use of PVS1. Caution should be used when interpreting relatively small multiexon deletions encoding regions of unknown function. There are currently no established guidelines regarding determination of clinically relevant transcripts. All types of variant interpretation would benefit from such guidance, but this falls outside the scope of this publication.
While applying PVS1 to multiexon deletions is reasonable, the pathogenicity of smaller, predicted in-frame deletions of one or two exons remains less certain. In these cases, reducing strength to strong may be appropriate if a deletion of the region is expected to impact protein function. Aligning with the rationale of the ACMG/AMP guidelines criteria, these modifications recommend increasing the strength of PM4 (i.e., PM4_Strong) to account for smaller, in-frame deletions that are not predicted to result in a functional null allele versus using PVS1_Strong. The moderate criteria PM4 for in-frame deletions in a nonrepeat region should be used if effect on function is unknown. In addition, classification of CNVs involving only the 3’ end of a gene requires special considerations as NMD would not be predicted (see PS1).
PS1
The PS1 criterion states that it is applicable for the “same amino acid change as a previously established pathogenic variant regardless of nucleotide change.”4 In line with this logic, PS1 could be applied to a deletion that is expected to have an equivalent effect at the level of the gene product as another known pathogenic variant (e.g., deletion involving the last coding exon when there is a truncating pathogenic variant with similar effect; predicted in-frame deletion when there is a smaller in-frame pathogenic deletion contained within the interval with similar effect).
PM2, BA1, BS1
Frequency criteria have significant impact on classifications; however, population databases are more robust for sequence variants than for CNVs partially due to limits of resolution in CNV databases. The DGV Gold Standard6 is among those most widely used by clinical laboratories for assessing CNV frequencies. Frequency-based criteria (PM2, BA1, and BS1) were modified to include DGV and, as available, gnomAD and/or laboratory internal data. The reliability of the newly released gnomAD structural variants (Collins RL, Brand H, Karczewski KJ, et al., An open resource of structural variation for medical and population genetics, Cold Spring Harbor Laboratory bioRxiv, 2019) should be established prior to clinical use since NGS structural variant calls may be more error-prone than simple sequence data. The DGV also has significant limitations, making the development of additional resources by testing laboratories, such as internally generated databases, critical. Similar to the ACMG/AMP guidelines for sequencing, no specific instruction or threshold was suggested for PM2, while BA1 had a threshold of 10× BS1 (Table S2). Table S2 recommends setting BS1 cut-offs by calculating the highest prevalence of all known phenotypes associated with the gene correlated to inheritance pattern and adjusted for other factors (e.g., penetrance). Examples of these frequency adjustments have been recently published by many groups.11,12
PM4
As described previously for PVS1, PM4 and PM4_Strong can be extrapolated to exon-level deletions. However, the ACMG/AMP guidelines lack criteria that would be applicable to an intragenic duplication detected by aCGH or multiplex ligation-dependent probe amplification (MLPA) as the location/orientation of the extra material is unknown. One breakpoint study of 119 gain CNVs demonstrated that 83% were tandem and direct and the majority of the remainder were interpreted as VUS because the effect could not be determined.13 This finding was confirmed by another study of breast cancer genes showing that duplications occurred in tandem 78% of the time with unknown status for the remainder.14 In contrast to tandem duplications involving an entire gene or either end of the gene, tandem intragenic duplications are likely to alter the gene product. For most genes, using PM4 for duplications not involving the 5’ and/or 3’ end (presumed intragenic) is recommended if consistent with the mechanism of disease. Rare exceptions are duplications where similar events have been interpreted as pathogenic, consistent with increased probability that the event is disrupting the gene for which these modifications support usage of PM4_Strong. PM4_Strong would be applicable to gene regions with a high frequency of published pathogenic duplications and/or with sequence data demonstrating tandem duplication as a recurrent disease mechanism. For example, 11% of disease-associated variants in the DMD gene are exonic duplications, and intragenic duplications are not observed in unaffected males.6,15 Intragenic duplication of DMD is a well-known mechanism of disease and the identification of an exon-level duplication not involving the ends of the gene in a patient with a dystrophinopathy is typically presumed damaging. Pathogenic criteria should not be used when the duplication includes the entire first or last coding exon(s) in the absence of additional data supporting gene disruption. Frame prediction is not required for similar reasons described above.
BP7
The definition of BP7 applies to synonymous variants with no predicted impact on the gene product. For CNVs, these adaptations extend the definition to include those involving noncanonical intronic or noncoding sequence not expected to impact the gene product based on size and proximity to potential regulatory sequences.
Validation of modified criteria across multiple laboratories
Figure 4 summarizes the results of testing three variant classification systems (rounds 1, 2, and 3) across independent teams from three different clinical genetic testing laboratories. Of the 12 variants from Table 1, seven were concordant in classification across all teams based on the lab-developed classification systems (round 1); two were discordant, but not at a medically significant level; and three variants had discordant classifications at the medically significant level: #8 (interlaboratory discordance) and #10 and #12 (inter- and intralaboratory discordance). However, when the same teams interpreted these 12 variants based on the ACMG/AMP guidelines4 (round 2), only two variants were concordant across all teams, seven were discordant but not medically significant, and three exhibited medically significant discordance: #1 (interlaboratory) and #10 and #12 (inter- and intralaboratory). Lastly, when all variants were interpreted according to the single-gene CNV modifications (round 3, Table S2), concordance was restored comparable with that of the lab-developed systems: six variants were completely concordant, four exhibited discordance that was not medically significant, and only two had medically significant discordance (inter- and intralaboratory): #10 and #12. A final replication study of 12 different CNVs showed very similar results (Fig. 4d).

Comparing tested classification systems across multiple laboratories. (a) Concordance of tested classification systems across multiple interpretation teams. Twelve variants were chosen to test across three different laboratories. Medically significant discordance is defined as LP/P versus VUS/LB/B. Y-axis is the total number of variants. (b) Variants exhibiting medically significant discordance per lab-developed systems (round 1). Y-axis is the total number of variant classifications. “Lab-developed” corresponds to round 1, “ACMG/AMP guidelines” corresponds to round 2, and “Modifications” corresponds to round 3. Y-axis is the total number of variants. (c) Classification of six pathogenic copy number variants (CNVs) using ACMG/AMP guidelines criteria before and after modifications. The six CNVs unanimously determined to be pathogenic using lab-developed systems (round 1) are shown here. Axes are labeled as in (b). (d) Concordance of classifications during the replication study of 12 different variants; graph is labeled as in (a). ACMG/AMP American College of Medical Genetics and Genomics/Association for Molecular Pathology, B benign, LB likely benign, LP likely pathogenic, P pathogenic, VUS variants of uncertain significance.
As illustrated in Fig. S2, without adequate criteria applicable to CNVs, the ACMG/AMP guidelines criteria in round 2 led participants to classify variants as LB ~1% of the time (1/84), VUS or LP each ~30% of the time (both 25/84), and P ~39% of the time (33/84). Overall, the adaptation of criteria allowed refinement of classifications to reflect our understandings of CNVs, resulting in less conservative VUS classifications and a more similar distribution to lab-developed systems (23 VUS in round 3 versus 21 VUS in round 1). There were fewer P classifications than in the lab-developed systems (44 vs. 55). Multiple reasons contributed, such as (1) deletions involving only the last exon of a gene require additional evidence to reach a P classification, and (2) these adjustments as well as the ACMG/AMP guidelines corrected an outlier classification of P by one laboratory for CNV #8 (Fig. 4b). Our study did not have the statistical power to determine if the CNV-specific adjustments were able to improve concordance for the TSC2 duplication. No concordance was reached with any system for the PRDM16 deletion because the discrepancy was due to different levels of familiarity with the gene and its disease mechanism among participants. A similar issue emerged around the MPZ gene during the replication study. These findings emphasize the need for guidelines regarding gene–disease correlations, which falls outside the scope of this publication.
Ease of use of CNV-specific criteria
In addition to improving concordance, another significant aim of these adjustments was ease of use. Due to lack of clear guidance for CNV classification by the ACMG/AMP guidelines, many participants found the unmodified system difficult to use (round 2). Six CNVs were unanimously classified P using lab-developed systems in round 1 (Fig. 4c, variants #2, #3, #5, #6, #9, and #11). For five of these, teams could not objectively call them pathogenic based on the ACMG/AMP guidelines alone. For example, multiple participants were hesitant to apply ACMG/AMP PVS1 or PM4-based criteria to the DMD presumed intragenic duplication (#9) due to uncertain location/orientation. In addition, multiple participants applied ACMG/AMP PVS1 and PM2 to the deletion of the entire CLN3 gene (#11) for an LP classification. However, modified PVS1 recommendations and modifications relevant to intragenic duplications improved objective criteria usage in round 3. All participants applied PM4_Strong to DMD duplication #9 and PVS1_Stand-alone to CLN3 deletion #11. For some difficult variants (e.g., deletions of the last exon only, #5 and #6), adjusted criteria provided limited utility.
Each of the ten participants in this study answered survey questions. After round 2, participants were asked what their overall experience was with the ACMG/AMP guidelines for this CNV exercise. Participants unanimously selected that the evidence criteria are not sufficient, and gave reasons such as insufficient consideration of duplications or intronic CNVs, lack of guidance for when similar but slightly different CNVs are known to be pathogenic, and lack of criteria to accurately classify some deletions that were considered clearly pathogenic. However, when asked about their overall experience with the modified criteria (round 3), 80% stated that they were easy to understand and apply (Table S4). In addition, when participants were asked how important it was for their laboratory that variant analysis can be done using one common classification system for sequence variants and CNVs in a single gene, 80% responded that this is “very important” and 20% indicated that this is “somewhat important.” Likewise, the same responses were given when users were asked “If a laboratory performs concurrent sequence analysis and deletion/duplication testing for a panel of genes, how important is it for you that all variant types included in the result report are interpreted using the same overall system?” (Table S4).
DISCUSSION
Single-gene CNVs vary in size and composition and consideration of these various types is necessary for the development of CNV classification systems. There is minimal published guidance for CNV classification. The distribution of consecutive CNVs observed during a six-month time window was similar to that previously reported by Truty et al.; however, direct comparison is not possible due to different panel/gene content studied and methodologies used.2
This publication aims to demonstrate how the ACMG/AMP guidelines can be extended to single-gene CNV classification by adapting several criteria and/or adjusting their strength. Using the adjusted criteria presented here (Table S2) yielded improved concordance of single-gene CNV classification compared with using only the ACMG/AMP guidelines and was user-friendly. The CNVs selected for testing represent diverse types and illustrate the challenges of CNV interpretation. For most types of single-gene CNVs, the modifications worked very well. Nevertheless, this study corroborates the need for a source of gene-level data for normal population CNVs to replace the less granular information from DGV.
Recently, the ClinGen Sequence Variant Interpretation (SVI) Working Group developed recommendations for PVS1 application to deletions/duplications involving exons within a single gene16 and tested these recommendations on 56 variants across ten genes. According to this SVI Working Group, “Whole gene deletions default to PVS1, assuming the gene in question meets the criterion for a LoF disease mechanism…. Although application of PVS1 (at a Very Strong level) would not reach a Pathogenic or Likely pathogenic classification using the combining rules in Richards et al.…, the SVI working group acknowledged that for a full gene deletion of a known haploinsufficient gene, a Pathogenic classification is warranted as long as there is no conflicting evidence….”16 PVS1_Stand-alone provides a solution.
While the recent ClinGen SVI Working Group recommendations pertaining to PVS1 usage discuss exon-level deletions and duplications, they do not provide guidance on other criteria, such as PS1. The SVI publication recommends using a moderate pathogenic criterion when an in-frame deletion involving a region of unknown function affects <10% of the protein but a strong criterion if >10% of the protein is involved.16 Establishing one size cut-off is difficult to apply broadly, therefore, no similar recommendation was made in this study. However, in principle the concepts are similar and the modifications tested here would allow PM4 application for an in-frame single-exon deletion of residues of unknown function with increasing strength for larger deletions. For deletions that are not predicted to result in NMD, the SVI Working Group recommends no higher than a strong criteria.16 However, this restricts the appropriate classification of P for a novel in-frame multiexon deletion involving a functionally crucial domain of an LOF gene in an affected individual. The modifications presented here recommend PVS1 + PM2 + PP4 and a P classification while the SVI group recommends PVS1_Strong instead of PVS1, which would lead to LP.
In addition, the modifications we tested apply a strong criterion for a presumed tandem intragenic duplication only when other similar known pathogenic duplications exist, suggesting that the gene/region is prone to pathogenic tandem duplication. For a novel duplication, presumed out-of-frame in an LOF gene with no known pathogenic gross duplications, PM4 and PM2 would apply, leading to a VUS classification. In contrast, the SVI group recommends using a strong criterion (PVS1_S) for a presumed tandem intragenic duplication in a patient involving an LOF gene provided that the product would be predicted to undergo NMD, otherwise no PVS1-based criterion is advised.16 This would allow for such a duplication to be classified as LP if PM2 also applies. The ACMG/AMP guidelines recommend that variants achieve an LP classification only when there is >90% certainty that the variant is pathogenic.4 However, for most genes there are insufficient studies to estimate the likelihood that a duplication observed by aCGH or MLPA is gene-disrupting. According to two reports, only 78–83% of studied duplications were proven to be in tandem and a gene-disrupting functional consequence could not be confidently predicted for 33/33 to 15/20 of the remaining duplications.13,14 Limitations prevent us from confidently extending the results of these studies to all exon-level duplications, including the limited number of unique CNVs/genes examined, not all the CNVs contained exonic sequence, and not all are single-gene CNVs. Consequently, for most genes, the fact that a duplication of intragenic material detected by copy number analysis is presumed tandem and absent from population databases may not be sufficient to classify it as LP (i.e., falls short of concluding the event is 90% likely to be gene-disrupting) without additional case-level criteria. Gene-level specifications apply for regions with intragenic duplications as a common disease mechanism. The modifications presented in this publication would recommend PM4_Strong + PM6 + PM2 for LP classification of an assumed de novo, novel intragenic DMD duplication overlapping known pathogenic duplications, presumed tandem and in-frame, identified in a patient with clinical fit. Based on the ACMG/AMP guidelines alone, multiple users in this study were hesitant to apply PM4 when chromosomal location and orientation were unknown; PM6 and PM2 criteria alone result in a VUS classification.
Applying the adjusted variant classification criteria presented here led to an increased concordance for the classification of single-gene CNVs compared with using only the ACMG/AMP guidelines. The two variants with medically significant discordance after interpretation using the modifications were similarly discordant using lab-developed systems. One of these variants, the TSC2 intragenic duplication, was discordant based on usage of nonmodified criterion PP1 (segregation) and PM4 versus PM4_Strong. The other variant involves a gene with a controversial disease mechanism.17 This underscores the utility of initiatives such as ClinGen’s Dosage Sensitivity Map and Gene–Disease Clinical Validity Curation.18,19 Discordance deemed not medically significant was also improved using this modified system. Although medical management is not likely affected by this type of discordance, reducing such conflict does improve overall confidence of reported classifications.
Since NGS technology has blurred the boundaries between sequence and copy number variants, it is timely to move toward developing a unified guideline for the interpretation of all types of DNA alterations. A single variant interpretation/classification system allows for reliability, efficiency, and clear, consistent clinical reports. As the clinical molecular genetics and cytogenetics fields are merging, laboratory geneticists should set their sights on a uniform interpretation system for all types of variants.
Change history
17 December 2019
The original version of this Article contained several typographical errors in Figure 4 and one in Figure 3. All these typos are deletions of “fi” (from definitively, significant, classification, and modifications). This has now been corrected in both the PDF and HTML versions of the Article.
17 December 2019
A Correction to this paper has been published: https://doi.org/10.1038/s41436-019-0725-5
References
Lindy As, Stosser MB, Butler E, et al. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia. 2018;59:1062–1071.
Truty R, Paul J, Kennemer M, et al. Prevalence and properties of intragenic copy number variation in Mendelian disease genes. Genet Med. 2018;21:114–123.
American Board of Medical Genetics and Genomics. Training Options. http://www.abmgg.org/pages/training_options.shtml. Accessed 10 April 2019.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Firth HV, Richards SM, Bevan AP, et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources. Am J Hum Genet. 2009;84:524–533.
MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42(D1):D986–92.
McGowan-Jordan J, Simons A, Schmid M. ISCN 2016: an international system for human cytogenomic nomenclature (2016). New York: Karger; 2016.
Harrison SM, Dolinsky JS, Knight Johnson AE, et al. Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genet Med. 2017;19:1096–1104.
Losson R, Lacroute F. Interference of nonsense mutations with eukaryotic messenger mRNA stability. Proc Natl Acad Sci U S A. 1979;76:5134–5137.
Maquat LE, Kinniburgh AJ, Rachmilewitz EA, Ross J. Unstable b-globin mRNA in mRNA-deficient b-thalassemia. Cell. 1981;27:543–553.
Kelly MA, Caleshu C, Morales A, et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet Med. 2018;20:351–359.
Gelb BD, Cavé H, Dillon MW, et al. ClinGen’s RASopathy Expert Panel consensus methods for variant interpretation. Genet Med. 2018;20:1334–1345.
Newman S, Hermetz KE, Weckselblatt B, Rudd MK. Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am J Hum Genet. 2015;96:208–220.
Richardson ME, Chong H, Mu W, et al. DNA breakpoint assay reveals a majority of gross duplications occur in tandem reducing VUS classifications in breast cancer predisposition genes. Genet Med. 2018;21:683–693.
Juan-Mateau J, Gonzalez-Quereda L, Rodriquez MJ, et al. DMD mutations in 576 dystrophinopathy families: a step forward in genotype-phenotype correlations. PLoS One. 2015;10:e0135189.
Abou Tayoun AN, Pesaran T, DiStefano MT, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018;39:1517–1524.
de Leeuw N, Houge G. Loss of PRDM16 is unlikely to cause cardiomyopathy in 1p36 deletion syndrome. Am J Hum Genet. 2014;94:153–154.
ClinGen. ClinGen dosage sensitivity map. https://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/. Accessed 10 April 2019.
ClinGen. Gene-disease clinical validity curation. https://www.clinicalgenome.org/curation-activities/gene-disease-validity/. Accessed 10 April 2019.
Lerner TJ, Boustany RN, Anderson JW, et al. Isolation of a novel gene underlying Batten disease, CLN3. The International Batten Disease Consortium. Cell. 1995;82:949–957.
Beggs AH, Hoffman EP, Snyder JR, et al. Exploring the molecular basis for variability among patients with Becker muscular dystrophy: dystrophin gene and protein studies. Am J Hum Genet. 1991;49:54–67.
Nakayama T, Ogiwara I, Ito K, et al. Deletions of SCN1A 5’ genomic region with promoter activity in Dravet syndrome. Hum Mut. 2010;31:820–829.
Sluiter MD, van Rensburg EJ. Large genomic rearrangements of the BRCA1 and BRCA2 genes: review of the literature and report of a novel BRCA1 mutation. Breast Cancer Res Treat. 2011;125:325–349.
Qiao Y, Tyson C, Hrynchak M, et al. Clinical application of 2.7M cytogenetics array for CNV detection in subjects with idiopathic autism and/or intellectual disability. Clin Genet. 2013;83:145–154.
Ashton EJ, Yau SC, Deans ZC, Abbs SJ. Simultaneous mutation scanning for gross deletions, duplications and point mutations in the DMD gene. Eur J Hum Genet. 2008;16:53–61.
OMIM. Baltimore, MD: Johns Hopkins University Center for Medical Genetics; 1996. https://omim.org/. Accessed 10 April 2019.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Dislcosure
T.B., L.M.S., D.A., D.T., H.M., H.C., H.G., J.M.M. and G.R. are full-time employees of GeneDx Inc., a wholly owned subsidiary of Bioreference Labs, which is a wholly owned subsidiary of OpkoHealth. While working on the manuscript, L.M.V., C.R. and A.L. were full-time employees of GeneDx Inc., a wholly owned subsidiary of Bioreference Labs, which is a wholly owned subsidiary of OpkoHealth. L.M.V. is currently employed at the Children’s National Health System Molecular Diagnostics Laboratory (Washington, DC). L.J.H.B. and A.A. are employees of EGL Genetic Diagnostics, LLC. A.K.J. is currently an employee and stockholder in Invitae Corporation. D.D.G. declares no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Brandt, T., Sack, L.M., Arjona, D. et al. Adapting ACMG/AMP sequence variant classification guidelines for single-gene copy number variants. Genet Med 22, 336–344 (2020). https://doi.org/10.1038/s41436-019-0655-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0655-2
Keywords
This article is cited by
-
Experience of copy number variation sequencing applied in spontaneous abortion
BMC Medical Genomics (2024)
-
Further evidence supporting the role of GTDC1 in glycine metabolism and neurodevelopmental disorders
European Journal of Human Genetics (2024)
-
A framework for the evaluation and reporting of incidental findings in clinical genomic testing
European Journal of Human Genetics (2024)
-
Intragenic homozygous duplication in HEPACAM is associated with megalencephalic leukoencephalopathy with subcortical cysts type 2A
neurogenetics (2024)
-
Pathogenic missense variation in PABPC1L/EPAB causes female infertility due to oocyte maturation arrest at the germinal vesicle stage
Journal of Assisted Reproduction and Genetics (2024)
