
Abstract
Purpose
The 22q11.2 deletion syndrome (22q11.2DS) is the most common microdeletion in humans, with highly variable phenotypic expression. Whereas congenital heart defects, palatal anomalies, immunodeficiency, hypoparathyroidism, and neuropsychiatric conditions are observed in over 50% of patients with 22q11DS, a subset of patients present with additional “atypical” findings such as craniosynostosis and anorectal malformations. Recently, pathogenic variants in the CDC45 (Cell Division Cycle protein 45) gene, located within the LCR22A–LCR22B region of chromosome 22q11.2, were noted to be involved in the pathogenesis of craniosynostosis.
Methods
We performed next-generation sequencing on DNA from 15 patients with 22q11.2DS and atypical phenotypic features such as craniosynostosis, short stature, skeletal differences, and anorectal malformations.
Results
We identified four novel rare nonsynonymous variants in CDC45 in 5/15 patients with 22q11.2DS and craniosynostosis and/or other atypical findings.
Conclusion
This study supports CDC45 as a causative gene in craniosynostosis, as well as a number of other anomalies. We suggest that this association results in a condition independent of Meier–Gorlin syndrome, perhaps representing a novel condition and/or a cause of features associated with Baller–Gerold syndrome. In addition, this work confirms that the phenotypic variability observed in a subset of patients with 22q11.2DS is due to pathogenic variants on the nondeleted chromosome.
Similar content being viewed by others


INTRODUCTION
The 22q11.2 deletion syndrome (22q11.2DS) is the most common microdeletion in humans, with an estimated prevalence of 1 in 2000–4000 live births and 1 in 1000 unselected fetuses.1
The 22q11.2 deletion is most frequently a de novo event that results from nonallelic homologous recombination between specific low copy repeats (LCRs), LCR22A and LCR22D, resulting in a ~3 MB deletion.2
The phenotypic expression of 22q11.2DS is highly variable. In general, the associated clinical findings include congenital cardiac abnormalities, palatal anomalies, and immunodeficiency in ~75% of patients; hypoparathyroidism and gastrointestinal differences in ~50%; genitourinary anomalies in ~30%. Furthermore, developmental delay, cognitive deficits, and psychiatric illness such as schizophrenia are important features of this condition. However, none of these features appear to be fully penetrant, and each exhibits variable expression. Moreover, a subset of patients presents with additional “atypical” findings, such as craniosynostosis, congenital diaphragmatic hernia, anorectal malformations, and airway anomalies.1
It has been previously suggested that large heterozygous deletions may unmask recessive variants on the remaining allele, resulting in pathogenic phenotypes. For example, it has already been demonstrated that, in patients with 22q11.2 deletion, hemizygous pathogenic variants in SNAP29 can unmask the autosomal recessive conditions CEDNIK (cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma) and potentially Kousseff syndromes.3 Likewise, hemizygous pathogenic variants in GP1BB cause Bernard–Soulier syndrome.4 Thus, there is increasing evidence that our ability to identify these pathogenic variants will help in understanding the function of previously uncharacterized genes that contribute to the unusual phenotypes observed in a subset of patients with 22q11.2DS. Conversely, and perhaps as importantly, it will have a great impact on understanding the causative genes underlying human autosomal recessive conditions.
Recently, pathogenic variants in the CDC45 (Cell Division Cycle protein 45) gene were suggested to be involved in the pathogenesis of both Meier–Gorlin syndrome (MGS) and craniosynostosis.5,6 MGS (MIM 224690) is a rare autosomal recessive primordial dwarfism disorder, characterized by microtia, short stature, and absent or hypoplastic patellae (Table 1).7 In 1998, Saha et al. mapped the CDC45 gene to chromosome 22q11.2, within the DiGeorge syndrome critical region (LCR22A–B).8
Here we identified CDC45 variants in a cohort of patients with a 22q11.2 deletion and craniosynostosis and/or other atypical developmental anomalies.
MATERIALS AND METHODS
As we had previously identified pathogenic variants on the remaining intact chromosome 22q11.2 allele in patients with atypical phenotypic features, such as Bernard–Soulier syndrome due to GP1BB pathogenic variants and polymicrogyria and keratoderma due to pathogenic variants in SNAP29, following the recent publication by Fenwick et al.5 we hypothesized that similar pathogenic variants in CDC45 gene might explain craniosynostosis in patients with 22q11.2DS. Thus, we retrospectively analyzed records on 1305 patients, from the 22q and You Center at The Children’s Hospital of Philadelphia (CHOP), a large comprehensive multidisciplinary program for patients with 22q11.2DS, founded following the identification of the N25 fluorescence in situ hybridization (FISH) probe at CHOP in the early 1990s. The study was approved by the Institutional Review Board of CHOP (07–005352) with appropriate informed consent obtained on all subjects. In addition, the study data were handled in compliance with the Health Insurance Portability and Accountability Act of 1996 (HIPAA) regulations. All patients had a laboratory confirmed 22q11.2 deletion and presented with “atypical” clinical features (found in <10% of overall cohort). These findings initially included craniofacial anomalies, such as craniosynostosis, choanal atresia or cleft lip, and limb differences, such as digitalized thumb or polydactyly. Eight patients with the aforementioned abnormalities and with available DNA met inclusion criteria for this study (Table S1). Based on the clinical presentation of these patients and on the recurrent phenotype of the patients with pathogenic variants in the CDC45 gene (Table 1), reported by Fenwick et al.,5 we further expanded the atypical findings spectrum, to include rare gastrointestinal anomalies, such as intestinal malrotation, imperforate/anteriorly displaced anus and congenital diaphragmatic hernia and short stature (in absence of any endocrine or metabolic cause) and patellar anomalies. Six additional patients were identified with these features and available DNA (Table S1).
Finally, in thinking about CDC45 as a putative cause for craniosynostosis in these patients we contacted authors who had previously reported such patients.9,10,11,12 The Children’s Hospital of Eastern Ontario and University of Ottawa (Canada) had DNA available for their previously reported patient9 (Table S1).
As the control group, we enrolled 85 patients with 22q11 deletion and without atypical clinical features described above, for whom exome sequencing data were received earlier. In addition, CDC45 targeted resequencing was performed in 48 more 22q11.2 patients without atypical findings.
Exome sequencing and variant analysis
Exome sequencing (ES) was performed on eight patients.
Because the initial hypothesis was that a pathogenic variant in the remaining allele causes the observed atypical findings, we focused only on the 22q11 region for ES data analysis.
Following genome library preparation using the TruSeq DNA Library Preparation Kit (Illumina), exome capture was performed with SeqCap EZ Human Exome Library v3.0 (Roche, NimbleGen) for eight patients (Table S1). Subsequently, 100-bp paired-end reads were generated on Illumina HiSeq2000 according to the manufacturer’s instructions. Reads were mapped to the reference human genome (hg19) using Burrows–Wheeler Aligner 0.7.8. The number of uniquely mapped reads ranged from 48 million to 108 million. The mean coverage at each target base varied from 54x to 139×, with a minimum of 80% of the bases covered by at least 30 reads in all the patients. SAMtools v 0.1.19 was used to sort and merge the data. Indel containing reads were realigned using RealignerTargetCreator and IndelRealigner of GATK 3.4.46. Variants were called using HaplotypeCaller, LeftAlignAndTrimVariants, and GenotypeGVCFs of GATK 3.5. Variants were annotated with ANNOVAR (version 2015) including for dbSNP (dbSNP137), 1000 Genomes data, and the ESP6500 panel (NHLBI GO Exome Sequencing Project [ESP], Seattle, WA), (http://evs.gs.washington.edu/EVS/). Functional predictions for the amino acid changes according to different models (SIFT, PolyPhen-2, LRT, and MutationTaster) were retrieved from dbNSFP (a database of human nonsynonymous single-nucleotide polymorphisms [SNPs] and their functional predictions).
All common variants with a minor allele frequency (MAF) >1% in either 1000 Genomes or ESP6500 were excluded. In addition, we filtered out nonexonic and non–splice site and synonymous variants. Variants were further filtered based on their frequency in ExAC (http://exac.broadinstitute.org/), gnomAD, and in NGSlogistics software.13 Finally, relevant variants were identified based on the available genotype–phenotype correlation and on known function of the affected genes.
Next-generation CDC45 targeted sequencing and Sanger sequencing
To screen seven more patients (Table S1) with a combination of 22q11.2 deletion and rare CDC45 variants, CDC45 exons, splice sites, and untranslated regions were sequenced. Except for exons 1, 2, 4, 17, and 18, all the exons had minimum coverage of 100 reads. A multiplex polymerase chain reaction (PCR) for joint amplification of 16 fragments encompassing all the exons, splice sites, and 3’ and 5’ UTRs was performed with tailed primers using Qiagen Multiplex PCR kit. Following PCR products purification, an indexing PCR was performed using Phusion High Fidelity kit. Subsequently, the PCR products were again purified, quantified, and pooled. Resulting libraries were sequenced on Illumina MiSeq platform according to the manufacturer’s recommendation for paired-end sequencing (2 × 300 bp) protocol. We used the same next-generation sequencing (NGS) analysis tools and adopted the same filtering method as for exome sequencing.
Due to their low coverage, exons 1, 2, 4, 17, and 18 were Sanger sequenced.
RESULTS
Rare hemizygous variants in the CDC45 gene were found in 5/15 (33%) patients with 22q11.2 deletion and atypical clinical features (3/7 [43%] patients with 22q11.2DS and craniosynostosis, 2 bicoronal, 1 metopic) and none of the 133 22q11.2DS patients from the control group (Fisher’s test p value <0.01 for a cut-off MAF <0.01). A synopsis of the clinical and genetic findings in these patients is provided in Table 2.
ES identified a missense variant (p.Arg283Cys) in the CDC45 gene in one patient (patient 1, Table 2). This variant is extremely rare in gnomAD and has been observed only at a heterozygous state. It affects a highly conserved amino acid and is predicted to be pathogenic by all in silico prediction tools.
CDC45 targeted sequencing identified rare nonsynonymous variants in four patients (patients 2–5, Table 2).
Case descriptions
Patient 1 (patient 3 from McDonald-McGinn et al.12 and patient 8 from Unolt et al.14), a female born via spontaneous vaginal delivery at 39 weeks gestation (WG) following an uneventful pregnancy, at birth experienced severe cardiorespiratory distress leading to the diagnosis of a complex congenital cardiac defect—truncus arteriosus (TA) and atrioventricular septal defect—and an anterior right-sided congenital diaphragmatic hernia (CDH). Additional clinical findings (Fig. 1) included low birth weight (1980 g, <1%), bicoronal craniosynostosis, hypertelorism with short palpebral fissures, microtia, left preaxial polydactyly with bilateral digitalized thumbs, anteriorly displaced anus (ADA), and a sacral dimple. Variational analysis of FGFR1, 2, 3, and TWIST1 were normal. Traditional cytogenetic studies initially revealed trisomy 8 mosaicism in 23% of lymphoblasts. In light of the TA, 22q11.2 deletion studies were performed using FISH and resulted positive. The patient underwent repair of CDH and congenital heart defect at age 7 and 13 days of life (DOL) respectively, succumbing to cardiac arrest 24 hours post cardiac repair. Postmortem exam revealed the following additional findings: a small abnormally shaped thyroid, absent parathyroid glands, absent thymus (not secondary to surgical removal), intestinal malrotation, an accessory spleen, left unicornuate uterus, and widespread white matter gliosis. Parental studies by FISH identified a 22q11.2 deletion in the mother whose medical history was significant for a congenital laryngeal anomaly and a learning disability.

Clinical features of patients with 22q11.2DS and CDC45 nonsynonymous variants: bicoronal craniosynostosis, hypertelorism with short palpebral fissures, microtia, and bilateral digitalized thumbs in patient 1 (a) and unicoronal craniosynostosis in patient 2 (b).
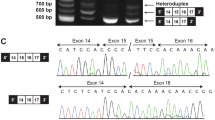
The CDC45 (p.Arg283Cys) variant was identified in the remaining allele using ES.
Patient 2, currently a 20-year-old female, presented for genetics consultation as part of the craniofacial clinic at CHOP at 16 months of age with a history of repaired unicoronal synostosis (Fig. 1) performed at an outside hospital (patient 2 from McDonald-McGinn et al.12), failure to thrive, gastroesophageal reflux disease, nasal regurgitation, and severe dysphagia requiring gastric tube placement. She was the former 2400 g (<2%) product of a 35-week pregnancy born to a G2P1–2 mother with a history of hypothyroidism. On physical exam at that time, she had thick, overfolded helices, evidence of asymmetric crying facies, a digitalized thumb, long slender fingers, laterally deviated great toes, overriding 2nd and 3rd toes, clinodactyly of the 5th toes bilaterally, and flat feet. Variational analysis for FGFR1, 2, 3, and TWIST1 were negative; however, the patient was found to have a 22q11.2 deletion by FISH. Parental studies were normal. Echocardiogram revealed aberrant left subclavian artery. She also has a history of mild hearing loss, chronic upper respiratory infections, mildly hypernasal speech, humoral deficits, and patellar subluxation. Height and head circumference were always below the curve: at 10 years height was 125 cm (<2%; 50% for 7.5 years) and occipitofrontal circumference (OFC) was 48.5 cm (<2%; 50% for 2 years of age). Developmentally, despite her learning difficulties she graduated high school. As a child her neuropsychological testing using the age appropriate Wechsler Intelligence Scale revealed a full-scale IQ of 85 with a verbal IQ of 91 and a performance IQ of 81.
Targeted sequencing of CDC45 revealed a c.241G>A variant.
Patient 3, a female, presented with a bilateral cleft lip and palate (Fig. 1), retrognathia, pseudoexotropia secondary to hypertelorism, short palpebral fissures, long slender fingers and toes, and ADA. Additional findings at birth included atrial septal defect, hypocalcemia, and T-cell lymphopenia. She was born at 37 WG to a G2P1 mother, via planned C-section due to a breech presentation. Pregnancy was uncomplicated. Birth weight was 3195 g (75–90%), length 51 cm (90%), OFC 34 cm (75–90%). Apgar scores were 7 and 8 at 1 and 5 minutes respectively. However, soon after birth she developed respiratory distress that required intubation and she was transferred to the neonatal intensive care unit (NICU) on DOL 3, where she underwent the first reconstructive surgery for her cleft lip and palate. At two weeks of age, FISH for 22q11.2 resulted positive. The patient is now 12 years old. Her height (50%) and weight (75%) are within normal limits. Additional findings include mild scoliosis, recurrent otitis media resulting in mild conductive hearing loss and requiring bilateral myringotomy tube placement, and a learning disability. Research multiplex ligation-dependent probe amplification (MLPA) studies revealed a standard LCR22A to LCR22D; however, in an attempt to understand her cleft lip and palate, research sequencing analysis of her remaining 22q11.2 allele initially revealed a hemizygous missense pathogenic variant of SNAP29, which has been associated with CEDNIK syndrome (patient 4 reported by McDonald-McGinn et al.3 and subsequently a CDC45 pathogenic variant [CDC45:NM_001178010:c.241G>A:p.(Val81Ile)]).
Patient 4, a male, was delivered at term weighing 2500 g (<3%) to a G5P4–5 mother. Pregnancy was unremarkable. Family history was contributory for his father having a “hole in his heart.” He was noted at birth to have imperforate anus and was transferred to a tertiary care facility for surgery on DOL 3. He had one seizure while in the hospital. No record of serum calcium level was available. Feeding difficulty necessitated gastric tube placement. A murmur was noted leading to diagnosis of TA requiring surgery at age 3 months. He was also noted to have hypospadias requiring surgical intervention and suffered from multiple urinary tract infections despite a normal voiding cystourethrogram. He was diagnosed with 22q11.2DS by FISH at CHOP on initial evaluation, at age 10 years, following the association of 22q11.2 deletions with conotruncal cardiac anomalies. Developmentally, he sat late, walked at 15 months, was able to vocalize words at 4 years, and spoke sentences at 5 years. He needed academic help especially in math and was in a self-contained class. On physical exam at age 10 years, his height was 128.5 cm (<5%, 50% for an 8-year-old), OFC was 51 cm (15%), helical rims were thickened and overfolded, the nasal tip was bulbous, tortuous retinal vessels and a submucosal cleft palate were noted along with a mild scoliosis, and fingers and toes were long and thin. He is currently 30 years of age and works in a supermarket.
Targeted sequencing of CDC45 revealed a (p.Ser183Cys) variant.
Patient 5 a male, previously reported by Al Hertani et al.9 was born at 37 WG to a 31-year-old multiparous mother, after a pregnancy complicated by prenatal diagnosis of cloverleaf skull, double outlet right ventricle, complex vascular ring, and a ventricular septal defect (at 23 WG). Family history was noncontributory. The infant was delivered by emergency cesarean for a nonreassuring fetal heart rate and required resuscitation and intubation. Birth weight was 2669 g (10%); length was at the 75th percentile. OFC could not be accurately assessed due to the severe cloverleaf skull. Radiographs of the skull revealed metopic and coronal craniosynostosis with wide patency of the entire sagittal suture (Fig. 1), later confirmed by skull radiographs. There was midfacial hypoplasia, a posterior cleft palate, and small mouth. He had sparse brows and lashes, upslanting and short palpebral fissures, proptosis, small pinnae, and attached lobes. Further findings on physical examination included an abdominal wall hernia above the umbilicus, cryptorchidism, a sacral dimple, ADA, arthrogryposis, proximal placement of the thumbs and tapering fingers, left 2–3 toe syndactyly, and bilateral aberrantly placed and crowded toes. Chest radiographs revealed a hook-shaped appearance to the distal clavicles. Laboratory tests revealed hypocalcemia and hypomagnesemia. The association of a conotruncal defect with hypocalcemia prompted FISH testing for 22q11.2 deletion, which was positive. SNP microarray confirmed a standard LCR22A–LCR22D 2.68-Mb deletion at 22q11.2. Plasma amino acids and urine organic acids were normal, as well as variational analysis of FGFR1, 2, 3, and TWIST1. Head computed tomography (CT) findings included near complete absence of the lateral orbital wall, shallow orbits, hypertelorism, and marked bitemporal protrusion. Magnetic resonance imaging (MRI) revealed severe enlargement of the temporal horns with thinning of the temporal lobe parenchyma, a small posterior fossa with compression of the cerebellum and brainstem, and herniation of the cerebellar tonsils below the foramen magnum. Thus, cranial vault decompression was performed on DOL 21 and three weeks later. Postoperatively the patient developed intracranial bleeds and seizures and then acquired Enterobacter cloacae pneumonia and became ventilator-dependent. He expired at 53 DOL.
Sanger sequencing revealed a c.36G>C variant in CDC45.
DISCUSSION
Herein we describe findings in five patients with 22q11.2 deletion syndrome (four identified via FISH and one with a standard LCR22A–LCR22D deletion identified via a SNP microarray) and rare nonsynonymous variants in the remaining allele of the CDC45 gene, including three patients with craniosynostosis. In the other two patients with rare missense CDC45 pathogenic variants, there was no evidence of cranial developmental anomalies. Interestingly, these patients too had important structural anomalies atypical for 22q11.2DS, most significantly some combination of anal/intestinal anomalies, limb differences, short stature, and craniofacial anomalies. These atypical findings are probably the result of CDC45 rare variants as they are associated with a significant excess of such variants.
The variant (p.Val81Ile) identified in patients 2 and 3 and the variant (p.Ser183Cys) identified in patient 4 have been reported in gnomAD, respectively in 25 and in 2 patients at homozygous state. Thus, one could argue that these variants are probably benign. However, taking into account the deletion of the second allele, the effect of these variants on the phenotype cannot be excluded. These variants are probably hypomorphic and the resulting phenotype could be correlated with the expression level and, subsequently, cause a more severe phenotype when they are in trans to a deletion than when homozygous. This has been observed in several diseases, including congenital vertebral malformations and Stargardt disease caused by the combination of null pathogenic variants with hypomorphic alleles in TBX6 and ABCA4 respectively.15,16 Additionally, the presence of homozygous variants in gnomAD could be explained by reduced penetrance in these presumably healthy individuals.
The pathogenicity of the c.36G>C variant in CDC45, identified in patient 5, is also difficult to predict. However it is already well known that the 3’ UTR is important for the stabilization, localization, and translation of messenger RNAs (mRNAs). A number of noncoding variants in regulatory regions have been previously reported as deleterious. For instance, variants in 3’ UTR have been reported as a cause of intellectual disability and congenital myasthenic syndrome.17,18 Although functional studies are needed to confirm its pathogenicity we report this variant because of the coexistence with atypical clinical findings in our patient. Additional evidence for the pathogenicity of the abovementioned variants is their lack in the control group of patients without atypical features.
The CDC45 protein is a cofactor within the eukaryotic replicative helicase, the so-called CMG complex, that consists of CDC45, MCM2–7 (minichromosome maintenance) proteins and GINS.19 Though in human cells its comprehensive functions during DNA replication are still not well understood, in yeasts and mice the Cdc45 protein is crucial for the initial DNA unwinding at replication origins, as well as for the elongation process.20,21,22,23,24 Thus, it is not surprising that the CDC45 gene is highly conserved among all eukaryotes and embryonic or neonatal lethality has been shown in Cdc45 knock-out mouse models.19,25,26 Interestingly, we found three patients with a nonsynonymous pathogenic variant on their only CDC45 allele, who survived to adolescence and even to adulthood and yet two succumbed to associated features. Notably for those three patients who survived the prediction algorithms were inconsistent (Table 2). Likewise, another recent paper reported on biallelic variants of CDC45 in 15 patients where 4 reached adolescence/adulthood.5 This suggests that either this gene is not as essential in humans as in mice, or, more likely, the phenotype depends on the position of the variant within the gene (Fig. 2). It is also possible that the diverse clinical picture is the result of the incomplete penetrance of the CDC45 pathogenic variants.25 However, to confirm this hypothesis it would be necessary to study the families/patients who have the pathogenic variants in the exact same position.

Schematic representation of CDC45 gene and localization of the identified pathogenic variants. Intronic regions are not drawn to scale. CDS coding DNA sequence, UTR untranslated region.
Pathogenic variants in CDC45 have not previously been associated with a specific phenotype in humans, until in 2016 Fenwick et al.5 identified biallelic pathogenic variants in the CDC45 gene in patients with a recurrent phenotype (Table 1) they reported to be consistent with Meier–Gorlin syndrome (MGS, MIM 224690), a rare autosomal recessive primordial dwarfism disorder, characterized by microtia, short stature, and absent or hypoplastic patellae.7 However, short stature and absent/hypoplastic patella were present in only 7/15 and 8/15 of the subjects, respectively. In addition, 11/15 of those patients with biallellic CDC45 pathogenic variants had craniosynostosis, which had not previously been reported as a feature of MGS. Based on these findings, the authors suggested that features associated with MGS should be expanded to include craniosynostosis.5 In our cohort with a 22q11.2 deletion and a CDC45 pathogenic variants, much like the Fenwick cohort, only one patient had patellar hypoplasia/agenesis, two of the three living patients had short stature, and three had craniosynostosis (Table 2). In turn, we suggest that the autosomal recessive condition associated with CDC45 biallelic pathogenic variants or monoallelic CDC45 pathogenic variants in the face of a chromosome 22q11.2 deletion should not be included in the MGS phenotypic spectrum but rather should be considered a novel condition. In considering the differential diagnosis for a best fit to account for the phenotype of our patients where craniosynostosis, gastrointestinal/anorectal malformations, and skeletal anomalies are key features, the diagnoses of Baller–Gerold syndrome (BGS, MIM 218600) and RAPADILINO syndrome (MIM 266280) appear to be a closer match (Table 1). Both these conditions are associated with biallelic pathogenic variants in the RECQL4 gene (RecQ like helicase 4, MIM 603780),27,28,29 which encode a protein also required for DNA replication initiation and progression.30 In particular, BGS (Table 1) is characterized by craniosynostosis; gastrointestinal/anorectal anomalies, such as imperforate/anteriorly placed anus; skeletal anomalies like radial ray defects; and short stature.31 However, BGS is only rarely associated with congenital heart defects, which are usually ventricular and/or atrial septal defects, and there is no correlation with cleft lip/palate and urogenital anomalies as we observed in our cohort. Thus, we suggest that a hemizygous pathogenic variant in CDC45 in association with a 22q11.2 deletion and/or biallelic pathogenic variants in CDC45 result in a novel phenotype. Specifically, a condition that is characterized by craniosynostosis, gastrointestinal differences including anorectal malformations, dysphagia, intestinal malrotation, and diaphragmatic and abdominal wall hernia, short stature, skeletal differences including upper limb anomalies, cleft lip/palate, congenital heart defects, genitourinary malformations in both male and females, and developmental differences. Perhaps the acronym CGS (C—craniosynostosis, cleft lip/palate; G—gastrointestinal and genitourinary; S—skeletal and short stature) syndrome would be more appropriate.
Disruption of the function of CDC45 in the replication initiation has already been proposed to cause impaired cell division during periods of rapid proliferation.5,32 When cell proliferation is disrupted during development of bone and cartilage this may lead to impaired growth (short stature), bone aplasia/hypoplasia (e.g., patellar or radial aplasia/hypoplasia), and premature fusion of cranial sutures (craniosynostosis). Moreover, consistent with its function in DNA replication, CDC45 expression has been suggested to be somehow involved in genomic instability and cancer proliferation.32,33,34 Also the RECQL4 gene has been reported to cause syndromes associated with increased malignancy risk.28,30 In our cohort and in the cohort of patients with CDC45 biallelic pathogenic variants5 no patient who reached adolescence/adulthood developed cancer. However, further follow-up is needed and it may be useful to extend the CDC45 testing to patients with 22q11.2 deletion syndrome and malignancies.
In conclusion, our study supports CDC45 as a causative gene in craniosynostosis and provides a genetic explanation for an atypical phenotype observed in a subset of patients with 22q11.2DS. Importantly, we suggest that a pathogenic variant in CDC45 should now be considered in every patient with a 22q11.2 deletion who presents with the following findings: craniosynostosis, anorectal anomalies/intestinal malrotation, short stature, upper limb anomalies, and cleft lip and palate. In addition, this observation demonstrates once again the utility of sequencing the remaining allele in patients with 22q11DS and atypical features.
References
McDonald-McGinn DM, Sullivan KE, Marino B, et al. 22q11.2 deletion syndrome. Nat Rev Dis Primers. 2015;1:15071.
Saitta SC, Harris SE, Gaeth AP, et al. Aberrant interchromosomal exchanges are the predominant cause of the 22q11.2 deletion. Hum Mol Genet. 2004;13:417–428.
McDonald-McGinn DM, Fahiminiya S, Revil T, et al. Hemizygous mutations in SNAP29 unmask autosomal recessive conditions and contribute to atypical findings in patients with 22q11.2DS. J Med Genet. 2013;50:80–90.
Kunishima S, Imai T, Kobayashi R, Kato M, Ogawa S, Saito H. Bernard–Soulier syndrome caused by a hemizygous GPIbβ mutation and 22q11.2 deletion. Pediatr Int. 2013;55:434–437.
Fenwick AL, Kliszczak M, Cooper F, et al. Mutations in CDC45, encoding an essential component of the pre-initiation complex, cause Meier–Gorlin syndrome and craniosynostosis. Am J Hum Genet. 2016;99:125–138.
Miller KA, Twigg SR, McGowan SJ, et al. Diagnostic value of exome and whole genome sequencing in craniosynostosis. J Med Genet. 2017;54:260–268.
de Munnik SA, Hoefsloot EH, Roukema J, et al. Meier–Gorlin syndrome. Orphanet J Rare Dis. 2015;10:114-015–0322-x.
Saha P, Thome KC, Yamaguchi R, Hou Z, Weremowicz S, Dutta A. The human homolog of Saccharomyces cerevisiae CDC45. J Biol Chem. 1998;273:18205–18209.
Al-Hertani W, Hastings VA, McGowan-Jordan J, Hurteau J, Graham GE. Severe craniosynostosis in an infant with deletion 22q11.2 syndrome. Am J Med Genet A. 2013;161A:153–157.
Rojnueangnit K, Robin NH. Craniosynostosis and radial ray defect: a rare presentation of 22q11.2 deletion syndrome. Am J Med Genet A. 2013;161A:2024–2026.
Yamamoto T, Sameshima K, Sekido K, et al. Trigonocephaly in a boy with paternally inherited deletion 22q11.2 syndrome. Am J Med Genet A. 2006;140:1302–1304.
McDonald-McGinn DM, Gripp KW, Kirschner RE, et al. Craniosynostosis: another feature of the 22q11.2 deletion syndrome. Am J Med Genet A. 2005;136A:358–362.
Ardeshirdavani A, Souche E, Dehaspe L, Van Houdt J, Vermeesch JR, Moreau Y. NGS-Logistics: federated analysis of NGS sequence variants across multiple locations. Genome Med. 2014;6:71–014-0071-9. eCollection 2014
Unolt M, DiCairano L, Schlechtweg K, et al. Congenital diaphragmatic hernia in 22q11.2 deletion syndrome. Am J Med Genet A. 2017;173:135–142.
Yang N, Wu N, Zhang L, et al. TBX6 compound inheritance leads to congenital vertebral malformations in humans and mice. Hum Mol Genet. 2019;28:539–547.
Zernant J, Lee W, Nagasaki T, et al. Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Cold Spring Harb Mol Case Stud. 2018;4:a002733.
Devanna P, van de Vorst M, Pfundt R, Gilissen C, Vernes SC. Genome-wide investigation of an ID cohort reveals de novo 3’UTR variants affecting gene expression. Hum Genet. 2018;137:717–721.
Dusl M, Senderek J, Muller JS, et al. A 3’-UTR mutation creates a microRNA target site in the GFPT1 gene of patients with congenital myasthenic syndrome. Hum Mol Genet. 2015;24:3418–3426.
Makarova KS, Koonin EV, Kelman Z. TheCMG (CDC45/RecJ, MCM, GINS) complex is a conserved component of the DNA replication system in all archaea and eukaryotes. Biol Direct. 2012;7:7–6150-7-7.
Shaikh TH, Gottlieb S, Sellinger B, et al. Characterization of CDC45L: a gene in the 22q11.2 deletion region expressed during murine and human development. Mamm Genome. 1999;10:322–326.
Pacek M, Walter JC. A requirement for MCM7 and Cdc45 in chromosome unwinding during eukaryotic DNA replication. EMBO J. 2004;23:3667–3676.
Gerhardt J, Guler GD, Fanning E. Human DNA helicase B interacts with the replication initiation protein Cdc45 and facilitates Cdc45 binding onto chromatin. Exp Cell Res. 2015;334:283–293.
Kohler C, Koalick D, Fabricius A, et al. Cdc45 is limiting for replication initiation in humans. Cell Cycle. 2016;15:974–985.
Szambowska A, Tessmer I, Kursula P, et al. DNA binding properties of human Cdc45 suggest a function as molecular wedge for DNA unwinding. Nucleic Acids Res. 2014;42:2308–2319.
Hestand MS, Nowakowska BA, Vergaelen E, et al. A catalog of hemizygous variation in 127 22q11 deletion patients. Hum Genome Var. 2016;3:15065.
Yoshida K, Kuo F, George EL, Sharpe AH, Dutta A. Requirement of CDC45 for postimplantation mouse development. Mol Cell Biol. 2001;21:4598–4603.
Van Maldergem L, Siitonen HA, Jalkh N, et al. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller–Gerold syndrome caused by mutations in the RECQL4 gene. J Med Genet. 2006;43:148–152.
Siitonen HA, Sotkasiira J, Biervliet M, et al. The mutation spectrum in RECQL4 diseases. Eur J Hum Genet. 2009;17:151–158.
Piard J, Aral B, Vabres P, et al. Search for ReCQL4 mutations in 39 patients genotyped for suspected Rothmund-Thomson/Baller–Gerold syndromes. Clin Genet. 2015;87:244–251.
Sangrithi MN, Bernal JA, Madine M, et al. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell. 2005;121:887–898.
Van Maldergem L, Piard J, Larizza L, Wang LL. Baller–Gerold syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews. Seattle (WA): University of Washington; 1993.
Pollok S, Bauerschmidt C, Sanger J, Nasheuer HP, Grosse F. Human Cdc45 is a proliferation-associated antigen. FEBS J. 2007;274:3669–3684.
Tomita Y, Imai K, Senju S, et al. A novel tumor-associated antigen, cell division cycle 45-like can induce cytotoxic T-lymphocytes reactive to tumor cells. Cancer Sci. 2011;102:697–705.
Colnaghi R, Carpenter G, Volker M, O’Driscoll M. The consequences of structural genomic alterations in humans: genomic disorders, genomic instability and cancer. Semin Cell Dev Biol. 2011;22:875–885.
Acknowledgements
This work was made possible by a grant from the National Science Center (OPUS NCN 2015/17/B/NZ5/01357 to N.B.) and via grants from the National Institutes of Health (UO1-MH191719, U01 MH119737-01, and PO1-HD070454 to T.B.C., B.S.E., E.H.Z., and D.M.-M.). We thank Greet Peeters and the Genomics Core Leuven for sequencing the CDC45 gene.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Consents
We have received and archived written consents for participation in this study from every individual whose data are included.
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Unolt, M., Kammoun, M., Nowakowska, B. et al. Pathogenic variants in CDC45 on the remaining allele in patients with a chromosome 22q11.2 deletion result in a novel autosomal recessive condition. Genet Med 22, 326–335 (2020). https://doi.org/10.1038/s41436-019-0645-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0645-4
Keywords
This article is cited by
-
The expanding genetic and clinical landscape associated with Meier-Gorlin syndrome
European Journal of Human Genetics (2023)
-
22q11.2 deletion — a tiny piece leading to a big picture
Nature Reviews Disease Primers (2020)
