
Abstract
Purpose
We aimed to assess the clinical value of prenatal testing for cystic fibrosis (CF) and whether ethical considerations would affect endpoint selection.
Methods
To determine effectiveness, we conducted a systematic literature review whose protocol outlined search strategies across eight databases, study inclusion criteria, and prespecified literature screening, data extraction, and synthesis processes. We conducted a scoping search on ethical considerations.
Results
The genetic test showed good diagnostic performance. A change in clinical management was observed: termination of pregnancy (TOP) occurred in most cases where two pathogenic variants were identified in a fetus of carrier parents (158/167; 94.6%). The TOP rate was lower in pregnancies where CF was diagnosed after fetal echogenic bowel detection (~65%). TOP and caring for a child with CF were both associated with poor short-term parental psychological outcomes. Ethical analyses indicated that informed decisions should have been the main endpoint, rather than CF-affected births prevented.
Conclusion
CF testing leads to fewer CF-affected births. It is difficult to assess whether this means the test is valuable, since patients may not value TOP primarily in terms of maternal or fetal health outcomes, psychological or otherwise. The value of testing should arguably be measured in terms of improving patient autonomy rather than health.
Similar content being viewed by others


INTRODUCTION
Cystic fibrosis (CF) is caused by biallelic pathogenic variants in the cystic fibrosis transmembrane conductance regulator (CFTR) gene on chromosome 7.1 CF is characterized by chronic bacterial airway and sinus infection, pancreatic exocrine insufficiency leading to incomplete breakdown of fats, congenital bilateral absence of the vas deferens (CBAVD) in males (leading to infertility), and a high sweat chloride concentration.2 Progressive respiratory disease is the major cause of mortality and morbidity among young CF patients.
CF and other CFTR-related autosomal recessive disorders are usually found in people of Caucasian descent.3 The incidence of CF is greater than 1 in 2000 births in Scotland and Ireland, is approximately 1 in 3500 births in the United States, and is only 1 in 40,000–100,000 births in India.3 Almost 2000 different CFTR variants have been identified, though 85% of the disease burden is attributable to only a few variants.4 The most common pathogenic variant in the CFTR gene is a three–base pair deletion at position 508 resulting in the loss of phenylalanine (F508del). This variant accounts for ~70% of pathogenic CFTR variants globally, but the occurrence of F508del varies among different population groups.1 Most variants are either small or point deletions/insertions that are able to be detected by some panel-based CFTR tests, exon scanning–based CFTR genetic tests, and DNA sequencing.5
Prenatal testing and family cascade testing of pathogenic CFTR variants are expected to lower the incidence of CF over time as individuals are provided with information that may cause them to avoid pregnancy or terminate an affected pregnancy.3
In the case of prenatal CFTR testing, a fetus is typically only tested if it is considered to be at high risk of CF. This is the case when both parents are known carriers of at least one pathogenic CFTR variant, or when a fetal echogenic bowel (FEB) is detected during a second-trimester ultrasound. One study has estimated that 0.8–13.3% of fetuses with FEB will have CF.6 When both parents are carriers of pathogenic CFTR variants, there is a one-in-four chance (25%) of CF in the child, which is 625 times greater than the risk in the general population (0.04%).7 Prenatal CFTR testing is usually done through chorionic villus sampling or amniocentesis; however, clinical practice is currently shifting toward noninvasive prenatal testing for CF.8,9
The aim of this systematic review is to determine the effectiveness of prenatal genetic testing to diagnose pathogenic CFTR variants in pregnancies at high risk of CF. A key challenge in assessing a prenatal test lies in selecting appropriate endpoints to determine the clinical utility of the test. With this in mind, we also conduct a review of ethical considerations relating to genetic testing to support the interpretation of clinical findings. The ethics review synthesizes ethical arguments and analyses published in the literature. It is specific to genetic testing, and we apply its insights to prenatal testing for CF.
Earlier versions of the systematic review and ethics review were previously conducted by the authors in 2015 as part of a contracted assessment of the technology for the Australian Department of Health,10 to aid the Australian Government in making a policy decision regarding whether genetic testing for pathogenic variants in the CFTR gene ought to be publicly funded.10 This paper provides a more up-to-date systematic review and introduces additional discussion and considerations. Any opinions expressed belong to the authors or cited writers alone.
MATERIALS AND METHODS
Literature sources
Biomedical databases were searched from 1 January 1989 (the year the CFTR gene was identified) to 2015, updated on 26 February 2019, and included PubMed, Embase, the Cochrane Library, Web of Science, Current Contents, CINAHL, Scopus, and EconLit. Additional (gray) literature was sought (Supplementary Table S1). Search strategies used key text words and indexing terms (e.g., MeSH or EmTree) combined using Boolean logic (Supplementary Table S2).
Additional targeted searches were undertaken to determine the health implications of management changes, e.g., investigating the effects of termination of pregnancy (TOP) versus the effects of raising a child with CF. Key texts in medical ethics11,12 and web resources13,14,15 were sourced to help frame the ethics review (Supplementary Table S3).
Methodological approach
The clinical utility of a test is ideally determined by a trial of the test (direct evidence). Comparative studies that evaluate the outcomes of genetic testing and subsequent patient management versus patient management without genetic testing are needed as evidence of the effectiveness of the test. This type of evidence is rare.16 Where direct evidence is not available, a linked-evidence approach may be used to estimate the clinical utility of a test. This approach involves linking evidence from different parts of the test treatment pathway.17 The evidence must be transferrable between linkages, i.e., the patient population, setting, and test delivery should be the same across studies. We have systematically reviewed the evidence of the diagnostic performance of prenatal CFTR testing (with clinical diagnosis of CF as the reference standard), using the linked-evidence approach to determine whether testing would impact clinical management decisions. We then considered whether this change in management would likely impact physical and psychological health outcomes. Evidence was systematically acquired and selected according to predetermined criteria for each step of the linked analysis (Supplementary Tables S4–S8). A review protocol was developed in consultation with an expert panel consisting of two clinical geneticists, a CF physician, and a senior genetic counselor. It is publicly available.18
Using clinically diagnosed CF as the reference standard is problematic, as this would be determined after birth. This was not possible where TOP was performed, and therefore false positive results may have been missed.
Study selection
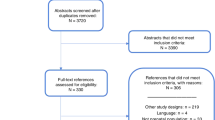
The study selection process is outlined in Fig. 1.19 Twenty percent of titles and abstracts were screened for eligibility by two reviewers (S.J.M.K. and S.N.). Since this achieved good agreement, the remainder were screened by one reviewer (S.J.M.K.) per AMSTAR 2.20 In cases of uncertainty regarding eligibility in the remainder, the second reviewer (S.N.) was consulted. Full-text articles were reviewed by S.J.M.K. The reference lists of potentially relevant studies were also searched.

Summary of the process used to identify and select studies for the systematic review.
Studies that met the prespecified criteria were eligible for inclusion (Supplementary Tables S4–S8). We excluded studies in languages other than English. The risk of bias in diagnostic performance studies was assessed using the QUADAS-2 tool.21 None of the included studies that assessed the impact of testing on management had a valid comparator, and this evidence was assessed using the Institute of Health Economics (IHE) appraisal checklist for case series.22
Study profiles were developed that described the authors, publication year, study design, level of evidence, risk of bias, population characteristics, setting, study location, the test and other relevant interventions, length of follow-up, possible comparators, and relevant outcomes.
The broad literature search on test effectiveness also returned numerous articles discussing ethics of prenatal genetic testing. These studies were selected for the ethics review.23,24,25,26,27
RESULTS
No direct evidence assessing the clinical utility of prenatal CFTR testing was identified.
Diagnostic performance of prenatal CFTR testing
Four studies investigated genetic CFTR testing in fetuses with two CF carrier parents. In these studies, biallelic variants in CFTR were detected in 8–22% of the fetuses, 38–58% were diagnosed as carriers, and 24–33% had no identifiable pathogenic variants28,29,30,31 (see Table 1). This roughly accords with the Mendelian inheritance pattern of a recessive trait (25% with the trait, 50% carrying the trait, 25% without the trait).
Two studies reported clinical outcomes after birth (the reference standard); no false negative test outcomes were recorded, i.e., there were no instances where children with CF were incorrectly identified as not having CF.29,30 However, neither the sensitivity nor specificity of testing could be determined, because carrier parents terminated pregnancies that were positive for biallelic pathogenic CFTR variants identified by prenatal tests.
Two studies investigated prenatal CFTR testing in pregnancies where FEB was detected. One study used denaturing gradient gel electrophoresis (DGGE) exon scanning of CFTR and DNA sequencing (see Table 1).30 However, no clinical outcomes were reported because there was no clinical reference standard to compare prenatal test accuracy against. The majority (46/49; 94%) had no detectable pathogenic variants and the remaining 6% (3/49) only had one pathogenic variant identified and were considered carriers. No fetus was found to have biallellic pathogenic CFTR variants; possibly due to the small sample size and the relatively low CF rate in pregnancies with FEB (2–13%). The second study tested 25 fetal samples in pregnancies with detected FEB.32 One pathogenic variant was detected (CF carrier) and no CF diagnoses occurred before or after birth.
Five studies reported test failure rates of various panel-based CFTR tests, though not all of these tests were prenatal.33,34,35,36,37 The median failure rate between seven different tests was 4.5% (range 0.0001–9). This suggests that diagnostic laboratories need to repeat ~4.5% of CFTR tests using panel-based tests. Test failures are usually resolved by sequencing the relevant amplicon or repeating the test.
No false positive results and very few false negative results were produced by pathogenic variant identification errors in a general population of patients suspected of CF (no prenatal testing). Amplification-refractory mutation system (ARMS) detection technology, used in two studies, produced the most identification errors due to the technology’s inability to distinguish between homozygous and heterozygous states. In these studies, the second pathogenic variant could not be determined in the homozygous samples (5% of samples).38,39
Misidentification errors were reported in two other noncomparative studies. In one study, 0.7% (7/1076) miscalls were made of the IVS8 5T/7T/9T polymorphism by a panel-based CFTR test using an oligonucleotide ligation assay.36 Whereas, a second study reported that the panel-based CFTR test using fragment analysis and fluorescent polymerase chain reaction (PCR) used in their laboratory failed to detect a one base pair difference and was unable to recognize the F508C variant from wild type.35
Change in management due to prenatal CFTR testing
Ten noncomparative studies reported the impact of prenatal CFTR testing on CF birth rates and TOP rates (see Tables 2 and 3).28,40,41,42,43,44,45,46,47,48 Almost all studies were judged to be of high (k = 5) or moderate (k = 4) quality, with one study rated as low quality, per the IHE checklist (Supplementary Table S9).22 Six and five studies included pregnant women with detected FEB, and where a high risk (25%) of CF in offspring was identified, respectively (one study included both groups). In the absence of a comparator group, i.e., assessing pregnancy outcomes where CFTR testing did not occur, we assumed that without prenatal CF testing the pregnancies would not be terminated. We recorded pregnancies terminated after a CF-positive test result, i.e., two pathogenic variants detected in the fetus, as a “change in management” due to testing. Subsequently, the rate of births with CF and “CF births avoided” were calculated.
The rate of TOP after detecting biallelic pathogenic variants in CFTR is shown in Tables 2 and 3. Where both parents were CF carriers (1:4 chance of having a child with CF) and two pathogenic variants were identified in the fetus, the pregnancy was terminated in 75–100% cases (158/167 [94.6%] pregnancies). The TOP rate was slightly lower in pregnancies where CF was diagnosed prenatally after FEB was detected (and parents were not known carriers). TOP occurred in 50/77 (65%) of these cases. Overall, the TOP rate ranged from 0 to 100% (median 69.4%) across studies.
In these high-risk populations a relatively low number of children were born with CF, due to the high TOP rate. This shows there is a management change, i.e., TOP, after a positive prenatal genetic test result in most cases, assuming that pregnancies would continue (i.e., no TOP) in the absence of genetic testing.
The effect of change in management due to CFTR testing: parental psychological health after TOP compared with raising a child with CF
No controlled studies investigated the health impact from TOP after a prenatal CF diagnosis compared with the impact of no testing (and likely no TOP) and caring for a child with CF. Therefore, we conducted an indirect, narrative synthesis of the psychological impacts of TOP following prenatal testing for fetal abnormalities compared with no testing and caring for a child with CF.
In the population undergoing TOP, post-traumatic stress,49,50,51,52,53,54,55,56 grief,53,57,58,59 anger and guilt,58 and depression50,52,54,59 were prevalent. Rates of grief (36–78%), post-traumatic stress (45.8–67%), and depression (~30%) were high in the first months after TOP, though these symptoms lessened over time. In these first few months post TOP, the rates of depression and anxiety were similar to those reported in studies among mothers of children with CF, with 48% and 20–34% of mothers scoring positive for anxiety and depression, respectively.60 The evidence showed that psychological health improved over time in women who experienced TOP for fetal abnormality; however, evidence is lacking whether this is also the case for mothers of a child with CF. Severe anxiety and depression in parents of a child with CF was associated with younger age of the child.60 However, another study reported that the age of affected children correlated with caregiver burden: the higher the age, the greater the burden.61 Men showed fewer psychological symptoms than women, both in couples undergoing TOP and couples parenting a child with CF.61
Ethics review
Prenatal genetic testing raises ethical issues regarding the aim of testing, informed choice and counseling, the disability rights critique, privacy and confidentiality, and risks versus benefits.
Aim of prenatal genetic testing
Prenatal CF genetic testing cannot be used to prevent or blunt the development of disease for an individual. It can only serve to inform parents of the presence of fetal abnormality or disease, while any subsequent TOP prevents the birth of an individual who has the abnormality or disease.62,63,64 Controversial appeals have been made for the availability of prenatal testing to reduce societal spending on high-cost medical conditions.62,65 Opponents emphasize that using prenatal testing to reduce the prevalence of diseases like CF is morally problematic, because this promotes TOP as a means of reducing the number of people with particular medical conditions or needs. A perspective known as the disability rights critique emphasizes some respects in which widespread selective TOP would be morally problematic (see below). A general view has emerged that prenatal genetic testing should be regarded purely as a means of providing pregnant women and couples with information and thereby enhancing their capacity to make an informed choice, when they regard that information as relevant. This aim accords with the principle of respect for autonomy.
It is possible that TOP reduces suffering for the woman or couple. In this article, we considered this by assessing parental psychological health after TOP compared with raising a child with CF. However, it is philosophically contentious to propose that TOP reduces suffering for the child. This is because it is arguably nonsensical to speak of the suffering or experience of an individual who is never born on account of TOP. It is also implausible to suggest that to live a CF-affected life is worse than to have never been born. (To suggest this also sends a discriminatory message about CF patients, as we discuss below under the disability rights critique.) A woman or couple may opt for TOP, then later achieve a subsequent pregnancy and have a child unaffected by CF. This child’s suffering has not been reduced, since this child is a different child (“nonidentical”) to the child whose birth was prevented by TOP. These matters are dealt with in philosophical discussion of the nonidentity problem.15
Informed choice and counseling
A person’s choice is autonomous when they understand all of the relevant information and make the choice voluntarily, free of manipulation and coercion. Nondirective counseling is therefore crucial before and after testing. If information is patchy or biased, or if counseling is directive, then the aim of providing information to enhance the capacity for autonomous choice is not met. Moreover, the aim effectively regresses to protecting society against the birth of children with often costly medical conditions through the use of selective TOP.63
It is plausible that women and couples experience some initial pressure to opt for testing simply by virtue of the offer coming from medical professionals and institutions, since these are vested with considerable power and social standing.65 This pressure potentially increases as the offer becomes more suggestive and as the opportunity to freely decline it becomes less clear, for instance through the routinization of testing.65 Moreover, the patient may experience the mere lawfulness and availability of TOP following a positive test result as sources of pressure toward testing if the patient interprets these in terms of societal expectation. All of this reinforces the importance of nondirective counseling.
Most commonly women cite the following reasons for changing their mind about testing:
-
“the level of risk … for the condition in question” (in this case, CF)
-
“the miscarriage risk of the test” (amniocentesis or chorionic villus sampling [CVS])
-
“the method of termination which would be offered if they chose to terminate the pregnancy” following a positive test result.62
It follows that these considerations are important to discuss during counseling, especially since women and health professionals tend to differ on what they regard as most important: women foremost value test safety, whereas health professionals foremost value test accuracy.66
Disability rights critique
The disability rights critique opposes selective TOP as a means of preventing the birth of individuals with a particular medical condition or need. Prenatal testing arguably “sends the message that it would have been better if those living with the targeted conditions had not been born.”64 Testing may therefore lead to increased discrimination within society, contrary to the principle of justice.45 Some people are concerned that prenatal testing will “degrade society’s willingness to accept and care for” children deemed abnormal, while simultaneously narrowing the range of acceptable normality.62 Furthermore, some writers have argued that widespread acceptance of selective TOP would lessen efforts to develop cures for genetic disorders, though they have not provided evidence of this occurring.62
Privacy and confidentiality
Genetic information is special in that genetic information about one individual can have implications for the lives of genetically related family members. For instance, if an individual opts against sharing their CF carrier status, this limits the ability of genetic relatives to make informed reproductive choices. An ethical dilemma arises for the health professional when conflicted between maintaining confidentiality and informing genetic relatives of their risk. Instead of providing nondirective counseling, counselors may seek to persuade patients to share their CF carrier status or ask patients to allow them to share that information.11 The potential benefits to genetic relatives could justify this. However, without authorization by the patient, disclosure by a health professional is ethically permissible (and may be required) only when the disclosure is both necessary and likely to succeed in lessening a serious threat to the health of a genetic relative, regardless of whether the threat is imminent.67 The Institute of Medicine’s Committee on Assessing Genetic Risks has proposed that the following criteria must be met to justify a breach of confidentiality: (1) “all attempts to elicit voluntary disclosure, including intensive genetic counseling, must have failed”; (2) the seriousness and likelihood of the harm posed by the pathogenic variants must be clear; and (3) an effective preventive or therapeutic intervention must be available.26 It is important to remember that there is only a risk for CF carriers and their genetic relatives if they are going to have children.
Prenatal testing can indicate nonpaternity.68 Pretest counseling should discuss this possibility to ensure that the choice whether to undertake testing is informed. The doctor can inform the family of the genetic discrepancy and indicate that presumably this was a rare case of a de novo variant, if this was a possibility.69 Unless the doctor has solid evidence, they have no right to suggest nonpaternity, since doing so could cause serious harm. If the doctor has solid evidence, then this should be shared with the patient if deemed relevant to medical decision making. Misinforming patients about test results would not be justified,68 but a doctor could justifiably judge that the finding of nonpaternity was secondary, outside the primary purpose of the test, and therefore not disclose it.
Weighing up risks and benefits
The principles of beneficence and nonmaleficence imply that the benefits of a genetic test ought to outweigh, perhaps substantially, the risks of harm. It is important to consider “the predictive value of the test,” the benefits and harms of interventions associated both with the test (e.g., amniocentesis and CVS) and “with a positive test result” (e.g., TOP), and “the availability and acceptability” of those interventions.70
DISCUSSION
The panel-based CFTR testing identified all variants that it is designed to detect.38,39 However, not every pathogenic CFTR variant is included in the panel, and a small percentage of patients with rarer variants would receive a false negative test result. Even if DNA sequencing were undertaken to try to detect these, around 2% would still be missed due to the inability to detect large deletions or insertions. Other methods such as multiplex ligation-dependent probe amplification (MLPA) would have to be conducted to identify these rarer variants. Moreover, the panel-based test failure rate was around 4.5%, so some tests would need to be repeated to get a valid result. It is important to explain to patients the likelihood of the fetus being truly negative in view of these limitations of the panel-based test.
Evidence indicating a change in patient management (e.g., TOP) following prenatal CFTR testing was found, although the data were noncomparative. TOP rates in pregnancies determined to be CF-positive after FEB detection varied widely (0–100%; Table 2). Heterogeneity among studies was expected in view of religious and cultural variation regarding the acceptability of TOP. However, all included studies were conducted in Europe, the UK, the United States, and Australia, which share religious and cultural histories. TOP is legal in these countries, with varying restrictions, though access to quality health care and TOP may vary. The most straightforward explanation of the difference in TOP rates may simply relate to chance, in connection with the very small sample size. The number of test positives was very low (n = 1 and n = 5) in studies with a 0% TOP rate. Furthermore, in the case of carrier couples, CFTR testing is usually done at a lower gestational age, compared with testing undertaken due to a second-trimester FEB detection. This means that carrier couples can opt for TOP at an earlier time point. This could explain why the TOP rate was higher in carrier couples (around 95% after a positive prenatal genetic test result) compared with FEB pregnancies (around 65% after a positive prenatal genetic test result). We did not have access to individual patient data and therefore were unable to conduct analyses adjusting for gestational age.
In our analysis of psychological outcomes, we found that short-term anxiety and depression rates were similar between women who underwent TOP due to fetal abnormality and women who had a child with CF (although evidence was noncomparative). Less is known about the long-term effects and other potential health impacts. Grief, post-traumatic stress, anger, depression, and guilt were prevalent after TOP. However, this decreased slowly over time. Similar rates of depression and anxiety were also prevalent among mothers of children with CF, with the rate of depression being associated with the younger age of the child, though caregiver burden increases with the age of the child. One study reported that only 8% and 10% of women who underwent TOP later reported feelings of regret and doubt respectively, suggesting that most women who opted for TOP valued having the option.54 It should be noted that the psychological health of parents of a child with CF may not be directly comparable with the psychological health of parents who underwent TOP for fetal abnormality, since fetal abnormalities include disorders that are substantially more severe than CF.
There are concerns that prenatal testing may degrade society’s willingness to accept and care for children with CF. This is one important reason why the success of testing ought not to be gauged in terms of the number of CF births prevented. Prenatal testing for fetal abnormalities should instead serve the aim of providing pregnant women and couples with information to enhance their autonomy. Nondirective counseling is crucial both for ensuring informed and voluntary consent to testing and for retaining the test’s aim of simply informing, rather than directing, reproductive choices. The exception to this is when the woman or couple should be counseled toward sharing information with family members who may attach significance to it when making their own reproductive decisions.
It is important not to confuse informing a choice with improving health, since a more informed choice may not lead to improved health. However, to assess the effectiveness of a health technology is to assess the benefit or value of the technology.71 While this typically lies in improving health, there are some exceptions, where the benefit or value lies elsewhere. For example, in vitro fertilization arguably does not improve health but instead overcomes the problem of childlessness. Our review of ethical considerations reveals persuasive reasons for regarding prenatal genetic testing for CF as a case where the benefit of the technology lies not in improving health, but in informing reproductive choice. In this way, our review suggests that test effectiveness ought to be assessed in terms of enhancing autonomy. Emerging technologies that can be used after birth to improve health outcomes for some CF patients, such as ivacaftor, may “influence the decision-making process of parents who receive a fetal diagnosis of CF,” leading to less TOP.72 This does not alter the effectiveness of CFTR testing if that is assessed in terms of enhancing autonomy rather than in terms of reducing the CF birth rate. By contrast, if technologies emerge that can be used antenatally to improve health outcomes for some CF-affected fetuses, then a codependent assessment of the effectiveness of CFTR testing may be warranted as part of new clinical pathways, including antenatal treatments, to prevent or slow the development of disease in affected individuals.
The ethics review also has implications for assessing the cost-effectiveness of CFTR testing, favoring as the primary endpoint something like cost per informed choice rather than cost per CF birth averted. One could measure the cost-effectiveness of CF testing followed by TOP in terms of health benefits for the parents. However, as we have shown, the evidence suggests that parental psychological health after TOP, compared with raising a child with CF, may be comparable, so there may be no clear health benefit for the parent. Meanwhile, one cannot measure the cost-effectiveness of CF testing followed by TOP in terms of health benefits for the fetus or child, since there are philosophical problems with describing TOP as beneficial for the fetus or any subsequent child.
This review was limited by the nonsystematic search of the ethics literature. Furthermore, we relied on a linked-evidence approach due to the absence of direct evidence. Psychological impact evidence was noncomparative and was therefore discussed as a naïve indirect comparison.
CONCLUSION
CFTR testing does influence patient management, very often leading to TOP when the test result is positive. Psychological outcomes for women undertaking TOP may be similar to psychological outcomes for women having a child with CF, at least in the short term, so the health impact of the test may be minimal in this respect. Two points are important to make.
First, the evidence is limited; the psychological impact of prenatal testing, and any subsequent decisions, requires further research. This may allow more value to be placed on the test. Change in management following a positive test result could also take the form of professional counseling in preparation for raising a child with CF, and the value of this should be assessable in terms of improved psychological outcomes for parents.
Second, the value of the test is difficult to assess because the value of TOP to women and couples opting for it may not be reducible to health outcomes, psychological or otherwise. It could be argued that CFTR testing ought to be valued in terms of enhancing patient autonomy rather than improving health.
References
Ratjen F, Doring G. Cystic fibrosis. Lancet. 2003;361:681–689.
Knowles MR, Durie PR. What is cystic fibrosis? N Engl J Med. 2002;347:439–442.
World Health Organisation. The molecular genetic epidemiology of cystic fibrosis: report of a joint meeting of WHO/ECFTN/ICF(M)A/ECFS. Genoa, Italy: World Health Organisation; 2004.
Egan ME. Genetics of cystic fibrosis: clinical implications. Clin Chest Med. 2016;37:9–16.
Moskowitz SM, Chmiel JF, Sternen DL, et al. Clinical practice and genetic counseling for cystic fibrosis and CFTR-related disorders. Genet Med. 2008;10:851–868.
De Oronzo MA. Hyperechogenic fetal bowel: an ultrasonographic marker for adverse fetal and neonatal outcome? J Prenat Med. 2011;5:9–13.
Welsh M, Ramsey B, Accurso F, Cutting G Cystic fibrosis. In: Scriver CR BA, Sly WS, Valle D, Childs B, Vogelstein B The Metabolic and Molecular Basis of Inherited Disease. 8th ed. New York: McGraw-Hill Inc; 2001.
Guissart C, Dubucs C, Raynal C, et al. Non-invasive prenatal diagnosis (NIPD) of cystic fibrosis: an optimized protocol using MEMO fluorescent PCR to detect the p.Phe508del mutation. J Cyst Fibros. 2017;16:198–206.
Hill M, Compton C, Karunaratna M, Lewis C, Chitty L. Client views and attitudes to noninvasive prenatal diagnosis for sickle cell disease, thalassaemia and cystic fibrosis. J Genet Couns. 2014;23:1012–1021.
Kessels S, Morona J, Mittal R, et al. Testing for hereditary mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. MSAC application 1216, Assessment Report. Canberra, ACT: Commonwealth of Australia; 2015. http://www.msac.gov.au/internet/msac/publishing.nsf/Content/1216-public.
Beauchamp TL, Childress JF. Principles of biomedical ethics. 5th ed. New York: Oxford University Press; 2001.
Rogers WA, Braunack-Mayer AJ. Practical ethics for general practice. Oxford, UK: Oxford University Press; 2004.
ALRC. Essentially yours: the protection of human genetic information in Australia (ALRC Report 96). 2003. http://www.alrc.gov.au/publications/report-96. Accessed 3 February, 2015.
Andersen S, Caron L, Cleret de Langavant G, et al. INAHTA's Working Group On Handling Ethical Issues: Stockholm, Sweden: INAHTA; 2005.
Roberts MA. The nonidentity problem. In: Edward N. Zalta editor. The Stanford Encyclopedia of Philosophy Stanford, USA: Metaphysics Research Lab, Stanford University; 2019.
Ferrante di Ruffano L, Davenport C, Eisinga A, Hyde C, Deeks JJ. A capture-recapture analysis demonstrated that randomized controlled trials evaluating the impact of diagnostic tests on patient outcomes are rare. J Clin Epidemiol. 2012;65:282–287.
Merlin T, Lehman S, Hiller JE, Ryan P. The “linked evidence approach” to assess medical tests: a critical analysis. Int J Technol Assess Health Care. 2013;29:343–350.
Medical Services Advisory Committee. Protocol. 1216—Testing for hereditary mutations in the Cystic Fibrosis conductance Transmembrane Regulator (CFTR) gene. 2014. http://www.msac.gov.au/internet/msac/publishing.nsf/Content/1216-public. Accessed 5 November2018.
Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. PLoS Med. 2009;6:e1000100.
Shea BJ, Reeves BC, Wells G, et al. AMSTAR 2: a critical appraisal tool for systematic reviews that include randomised or non-randomised studies of healthcare interventions, or both. BMJ. 2017;358:j4008.
Whiting PF, Rutjes AW, Westwood ME, et al. QUADAS-2: a revised tool for the quality assessment of diagnostic accuracy studies. Ann Intern Med. 2011;155:529–536.
Moga C, Guo B, Schopflocher D, Harstall C. Development of a quality appraisal tool for case series studies using a modified Delphi technique. Alberta, Edmonton: Institute of Health Economics; 2012.
Hildt E. Autonomy and freedom of choice in prenatal genetic diagnosis. Med Health Care Philos. 2002;5:65–71.
Kinder BK. Genetic and biochemical screening for endocrine disease: II. Ethical issues. World J Surg. 1998;22:1208–1211.
Wilcken B. Ethical issues in genetics. J Paediatr Child Health. 2011;47:668–671.
Winslow ER, Kodner IJ, Dietz DW. Ethics and genetic testing. Semin Colon Rectal Surg. 2005;15:186–190.
Giarelli E. Ethical issues in genetic testing. The experiences of one family diagnosed with an inherited cancer syndrome. J Infus Nurs. 2001;24:301–310.
Castaldo G, Martinelli P, Massa C, et al. Prenatal diagnosis of cystic fibrosis: a case of twin pregnancy diagnosis and a review of 5 years' experience. Clin Chim Acta. 2000;298:121–133.
Collazo T, Lopez I, Clark Y, et al. Antenatal testing for cystic fibrosis in cuba, 1988-2011. MEDICC Rev. 2014;16:18–21.
Kanavakis E, Efthymiadou A, Strofalis S, Doudounakis S, Traeger-Synodinos J, Tzetis M. Cystic fibrosis in Greece: molecular diagnosis, haplotypes, prenatal diagnosis and carrier identification amongst high-risk individuals. Clin Genet. 2003;63:400–409.
Saker A, Benachi A, Bonnefont JP, et al. Genetic characterisation of circulating fetal cells allows noninvasive prenatal diagnosis of cystic fibrosis. Prenat Diagn. 2006;26:906–916.
Miller ME, Allen VM, Brock JAK. Incidence and carrier frequency of CFTR gene mutations in pregnancies with echogenic bowel in Nova Scotia and Prince Edward Island. J Obstet Gynaecol Can. 2018;40:896–902.
Axton RA, Brock DJH. A single-tube multiplex system for the simultaneous detection of 10 common cystic fibrosis mutations. Hum Mutat. 1995;5:260–262.
Edelmann L, Hashmi G, Song Y, Han Y, Kornreich R, Desnick RJ. Cystic fibrosis carrier screening: validation of a novel method using BeadChip technology. Genet Med. 2004;6:431–438.
Nagy B, Richárd Nagy G, Lázár L, Bán Z, Papp Z. Detection of ΔF508del using quantitative real-time PCR, comparison of the results obtained by fluorescent PCR. Fetal Diagn Ther. 2007;22:63–67.
Strom CM, Clark DD, Hantash FM, et al. Direct visualization of cystic fibrosis transmembrane regulator mutations in the clinical laboratory setting. Clin Chem. 2004;50:836–845.
Strom CM, Huang D, Chen C, et al. Extensive sequencing of the cystic fibrosis transmembrane regulator gene: assay validation and unexpected benefits of developing a comprehensive test. Genet Med. 2003;5:9–14.
Houdayer C, Cazeneuve C, Cougoureux E, et al. Clinical evaluation of the CF(12)m cystic fibrosis DNA diagnostic kit. Clin Chem. 1998;44:1346–1348.
Tomaiuolo R, Spina M, Castaldo G. Molecular diagnosis of cystic fibrosis: comparison of four analytical procedures. Clin Chem Lab Med. 2003;41:26–32.
Ameratunga DM, Said JM, Reidy K, Palma-Dias R. Perinatal outcomes following the ultrasound diagnosis of echogenic bowel: an Australian perspective. Fetal Diagn Ther. 2012;31:179–184.
De Becdelièvre A, Costa C, Jouannic JM, et al. Comprehensive description of CFTR genotypes and ultrasound patterns in 694 cases of fetal bowel anomalies: a revised strategy. Hum Genet. 2011;129:387–396.
Ghose I, Mason GC, Martinez D, et al. Hyperechogenic fetal bowel: a prospective analysis of sixty consecutive cases. Br J Obstet Gynaecol. 2000;107:426–429.
Muller F, Simon-Bouy B, Girodon E, Monnier N, Malinge MC, Serre JL. Predicting the risk of cystic fibrosis with abnormal ultrasound signs of fetal bowel: results of a French molecular collaborative study based on 641 prospective cases. Am J Med Genet. 2002;110:109–115.
Scotet V, Audrézet MP, Roussey M, et al. Impact of public health strategies on the birth prevalence of cystic fibrosis in Brittany, France. Hum Genet. 2003;113:280–285.
Scotet V, Duguépéroux I, Audrézet MP, et al. Prenatal diagnosis of cystic fibrosis: the 18-year experience of Brittany (western France). Prenat Diagn. 2008;28:197–202.
Slotnick RN, Abuhamad AZ. Prognostic implications of fetal echogenic bowel. Lancet. 1996;347:85–87.
Tomaiuolo R, Nardiello P, Martinelli P, Sacchetti L, Salvatore F, Castaldo G. Prenatal diagnosis of cystic fibrosis: an experience of 181 cases. Clin Chem Lab Med. 2013;51:2227–2232.
Archibald AD, Smith MJ, Burgess T, et al. Reproductive genetic carrier screening for cystic fibrosis, fragile X syndrome, and spinal muscular atrophy in Australia: outcomes of 12,000 tests. Genet Med. 2018;20:513–523.
Daugirdaite V, van den Akker O, Purewal S. Posttraumatic stress and posttraumatic stress disorder after termination of pregnancy and reproductive loss: a systematic review. J Pregnancy. 2015;2015:646345.
Davies V, Gledhill J, McFadyen A, Whitlow B, Economides D. Psychological outcome in women undergoing termination of pregnancy for ultrasound-detected fetal anomaly in the first and second trimesters: a pilot study. Ultrasound Obstet Gynecol. 2005;25:389–392.
Kersting A, Dorsch M, Kreulich C, et al. Trauma and grief 2-7 years after termination of pregnancy because of fetal anomalies—a pilot study. J Psychosom Obstet Gynaecol. 2005;26:9–14.
Kersting A, Kroker K, Steinhard J, et al. Psychological impact on women after second and third trimester termination of pregnancy due to fetal anomalies versus women after preterm birth-a 14-month follow up study. Arch Womens Ment Health. 2009;12:193–201.
Korenromp MJ, Christiaens GC, van den Bout J, et al. Long-term psychological consequences of pregnancy termination for fetal abnormality: a cross-sectional study. Prenat Diagn. 2005;25:253–260.
Korenromp MJ, Page-Christiaens GC, van den Bout J, et al. Psychological consequences of termination of pregnancy for fetal anomaly: similarities and differences between partners. Prenat Diagn. 2005;25:1226–1233.
Korenromp MJ, Page-Christiaens GC, van den Bout J, et al. A prospective study on parental coping 4 months after termination of pregnancy for fetal anomalies. Prenat Diagn. 2007;27:709–716.
Salvesen KA, Oyen L, Schmidt N, Malt UF, Eik-Nes SH. Comparison of long-term psychological responses of women after pregnancy termination due to fetal anomalies and after perinatal loss. Ultrasound Obstet Gynecol. 1997;9:80–85.
Geerinck-Vercammen CR, Kanhai HH. Coping with termination of pregnancy for fetal abnormality in a supportive environment. Prenat Diagn. 2003;23:543–548.
Iles S, Gath D. Psychiatric outcome of termination of pregnancy for foetal abnormality. Psychol Med. 1993;23:407–413.
Korenromp MJ, Page-Christiaens GC, van den Bout J, Mulder EJ, Visser GH. Adjustment to termination of pregnancy for fetal anomaly: a longitudinal study in women at 4, 8, and 16 months. Am J Obstet Gynecol. 2009;201:160 e161–167.
Quittner AL, Goldbeck L, Abbott J, et al. Prevalence of depression and anxiety in patients with cystic fibrosis and parent caregivers: results of the International Depression Epidemiological Study across nine countries. Thorax. 2014;69:1090–1097.
Fitzgerald C, George S, Somerville R, Linnane B, Fitzpatrick P. Caregiver burden of parents of young children with cystic fibrosis. J Cyst Fibros. 2018;17:125–131.
Aksoy S. Antenatal screening and its possible meaning from unborn baby's perspective. BMC Med Ethics. 2001;2:E3.
de Jong A, de Wert GM. Prenatal screening: an ethical agenda for the near future. Bioethics. 2015;29:46–55.
de Jong A, Dondorp WJ, Frints SGM, de Die-Smulders CEM, de Wert G. Advances in prenatal screening: the ethical dimension. Nat Rev Genet. 2011;12:657–663.
Munthe C. A new ethical landscape of prenatal testing: individualizing choice to serve autonomy and promote public health: a radical proposal. Bioethics. 2015;29:36–45.
De Jong A, Maya I, Van Lith JMM. Prenatal screening: current practice, new developments, ethical challenges. Bioethics. 2015;29:1–8.
Suthers GK, McCusker EA, Wake SA. Alerting genetic relatives to a risk of serious inherited disease without a patient's consent. Med J Aust. 2011;194:385–386.
Hill J. More than I wanted to know. Lancet. 2004;363:1654.
Campbell K. Disclosure of nonpaternity. Lancet. 2004;364:327. author reply 328
Burke W, Press N. Genetics as a tool to improve cancer outcomes: ethics and policy. Nat Rev Cancer. 2006;6:476–482.
HTA Glossary. Effectiveness. http://htaglossary.net/effectiveness. Accessed 13 March 2019.
Elsas CR, Schwind EL, Hercher L, Smith MJ, Young KG. Attitudes toward discussing approved and investigational treatments for cystic fibrosis in prenatal genetic counseling practice. J Genet Couns. 2017;26:63–71.
Acknowledgements
We thank Judy Morona for analyzing the diagnostic accuracy evidence and Thomas Vreugdenburg for critical revision of the manuscript. Earlier versions of the systematic review and ethics review were previously conducted in 2015 as part of an Australian Department of Health contracted assessment of the technology (Kessels S, Morona J, Mittal R, et al. Testing for hereditary mutations in the cystic fibrosis transmembrane conductance regulator [CFTR] gene. 2015. MSAC application 1216, Assessment Report, Commonwealth of Australia, Canberra, ACT). Any opinions expressed belong to the authors or cited writers alone.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Kessels, S.J.M., Carter, D., Ellery, B. et al. Prenatal genetic testing for cystic fibrosis: a systematic review of clinical effectiveness and an ethics review. Genet Med 22, 258–267 (2020). https://doi.org/10.1038/s41436-019-0641-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0641-8
This article is cited by
-
Addressing the dark matter of gene therapy: technical and ethical barriers to clinical application
Human Genetics (2022)
-
Fifty years of Shannon information theory in assessing the accuracy and agreement of diagnostic tests
Medical & Biological Engineering & Computing (2022)
-
The Core Outcome DEvelopment for Carrier Screening (CODECS) study: protocol for development of a core outcome set
Trials (2021)
