
Abstract
Purpose
Sifrim–Hitz–Weiss syndrome (SIHIWES) is a recently described multisystemic neurodevelopmental disorder caused by de novo variants in CHD4. In this study, we investigated the clinical spectrum of the disorder, genotype–phenotype correlations, and the effect of different missense variants on CHD4 function.
Methods
We collected clinical and molecular data from 32 individuals with mostly de novo variants in CHD4, identified through next-generation sequencing. We performed adenosine triphosphate (ATP) hydrolysis and nucleosome remodeling assays on variants from five different CHD4 domains.
Results
The majority of participants had global developmental delay, mild to moderate intellectual disability, brain anomalies, congenital heart defects, and dysmorphic features. Macrocephaly was a frequent but not universal finding. Additional common abnormalities included hypogonadism in males, skeletal and limb anomalies, hearing impairment, and ophthalmic abnormalities. The majority of variants were nontruncating and affected the SNF2-like region of the protein. We did not identify genotype–phenotype correlations based on the type or location of variants. Alterations in ATP hydrolysis and chromatin remodeling activities were observed in variants from different domains.
Conclusion
The CHD4-related syndrome is a multisystemic neurodevelopmental disorder. Missense substitutions in different protein domains alter CHD4 function in a variant-specific manner, but result in a similar phenotype in humans.
Similar content being viewed by others


INTRODUCTION
Sifrim–Hitz–Weiss syndrome (SIHIWES, MIM 617159) is a multisystemic neurodevelopmental disorder caused by heterozygous missense variants in chromodomain helicase DNA-binding protein 4 (CHD4).1,2 This condition is part of an increasingly recognized group of Mendelian disorders involving chromatin remodeling abnormalities. The initial characterization of SIHIWES was based on reverse phenotyping, i.e., we compared the clinical features of five individuals with de novo variants in CHD4 that underwent exome sequencing (ES) for developmental delay and congenital anomalies.1 We identified several similar features supporting a common etiology, and similar findings were found independently by a second group.2 All published cases had de novo missense variants but their effect on the enzymatic activity was not fully elucidated. Because the syndrome has only been defined recently as a clinical entity, there is scarce data regarding the clinical spectrum, natural history, and genotype–phenotype correlations—information required for counseling patients’ families and clinical management of the disorder. In this study we describe the clinical features, molecular findings, and genotype–phenotype correlations of 32 individuals with mostly de novo CHD4 variants. Furthermore, we provide functional data on the effect of variants in different domains on CHD4 protein activity.
CHD4 encodes an ATP-dependent chromatin remodeler, a core component of the nucleosome remodeling and histone deacetylation (NuRD) complex.3,4,5,6,7,8 This complex is widely expressed and acts mainly, but not exclusively, as a transcriptional repressor. CHD4, as well as CHD3 and CHD5, belong to the CHD subfamily II, which is characterized by the presence of PHD fingers and chromodomains, in addition to the SNF2-like ATPase/helicase core.5,8,9 These three paralogs incorporate into the NuRD complex in a mutually exclusive manner,10,11 and each enzyme was found to have distinct roles in the cortical development of mice.11 Similarly to CHD4, de novo variants in CHD3 are associated with a neurodevelopmental syndrome.12 To date, there are no reports of pathogenic germline variants in CHD5 in patients with intellectual disability (ID), but it is a known tumor suppressor gene expressed mainly in the nervous system and frequently deleted in neuroblastoma.13
The ATPase and helicase domains of CHD4 resemble SNF2, the catalytic subunit of the SWI/SNF complex in yeast, and provide the energy for nucleosome sliding.9 The PHD and chromodomains were found to regulate ATPase and chromatin remodeling activity by binding nucleosome and histone H3 tails.14,15 In addition, there are several highly conserved domains in the N-terminal and C-terminal regions of CHD4, some of which have not been previously investigated. Here, we demonstrate that de novo missense substitutions in different domains of CHD4 result in a similar phenotype in humans. Furthermore, our data, together with previous publications, show missense substitutions in different domains affect adenosine triphosphate (ATP) hydrolysis and chromatin remodeling in a variant-specific manner.
MATERIALS AND METHODS
Patients
Since the initial publications in 2016, we reached out to additional clinicians following individuals with de novo variants in CHD4. Connections were made through the Deciphering Developmental Disorders (DDD) group in the UK, GeneDx (Gaithersburg, MD), the Pediatric Cardiac Genomic Consortium (PCGC), and different clinicians who contacted us directly. The clinician of each participant provided clinical data including information on growth and development, physical examination, and laboratory and imaging studies. In addition, data were provided on the detected variant. The majority of participants had clinical exome sequencing and the others underwent next-generation sequencing panels and exome sequencing through various research projects (DDD, PCGC). All participants had a nondiagnostic chromosomal microarray prior to sequencing. All the participants or their families consented to participation in the study. Informed consent was obtained for the publication of all photographs. The study was approved by the National Human Genome Research Institute institutional review board (IRB) (clinicaltrials.gov: NCT01294345).
Protein modeling
There is no experimental structure of the entire CHD4 protein; only the N-terminal (CHDNT, PDB #2N5N), PHD1 (#2L5U), PHD2 (#2L75), and chromodomains (#4O9I) structures have been solved. We modeled both PHD1–2 domains, and the chromodomains together with the SNF2-like region (based on the previously solved CHD1, #3MWY) using SWISS-MODEL.16 Both domains of unknown function (DUF1087 and DUF1086) and the C-terminal domain (CHDCT2) were modeled using I-TASSER;17 the best models were chosen for presentation according to their c-scores. The 3D locations of CHD4 substitutions were mapped on the corresponding domain structures.
ATPase and chromatin remodeling assays
A detailed description of the methods is provided in the Supplementary note methods. The CHD4 protein was prepared as previously described,12 and nucleosome reconstitution was performed as previously described18,19
ATPase assay
Reaction mixtures (10 μL) of 20 mM Tris-HCl pH 7.5, 1 mM MgCl2, 1 mM DTT, and 0.1 mg/mL BSA were prepared. As a tracer, 2 μL of γ-32P ATP was added to 20 μL of 1 mM ATP, which was then added to the reaction mixture at a final concentration of 100 μM ATP. Nucleosome (12 nM) or naked 347-bp DNA (12 nM) was added to the mixture, and the reaction was initiated by the addition of wild-type or mutant CHD4 protein (at a final concentration of 25 nM). Samples were incubated at 37 °C for 40 minutes; the addition of 2 μL of 0.5 M EDTA terminated the reaction. Aliquots (2.5 μl) of each reaction mixture were spotted onto a PEI-cellulose thin-layer chromatography (TLC) plate and placed into a mobile phase of 1 M formic acid and 0.5 M LiCl and allowed to develop for 60 minutes. TLC plates were exposed to a phosphorimager overnight and ATP hydrolysis was quantified using Image Quant Software. Three individual experiments from two biological replicates (n = 6) were conducted for each mutant.
Chromatin remodeling assay
A restriction enzyme accessibility assay was used to measure the remodeling activities.19 Each reaction took place in the presence of 20 mM Tris-HCl pH 7.5, 1 mM MgCl2, 1 mM DTT, 0.1 mg/mL BSA, 1 mM ATP, and 5 U HhaI. Wild-type or mutant CHD4 was added to 12 nM nucleosomes at final concentrations of 12 nM, 6 nM, and 3 nM to initiate the reaction. Mixtures were incubated at 37 °C for 60 minutes and quenched by the addition of 2 μL of proteinase K buffer (167 mM EDTA, 1.7% SDS, and 6.7 mg/mL proteinase K), then incubated at 50 °C for 15 minutes for deproteination to take place. Samples were analyzed by 6% native polyacrylamide gel electrophoresis. DNA fragments were visualized on a ChemiDox XRS system (Bio-Rad) and were quantitated using Image Lab software. Three individual experiments from two biological replicates (n = 6) were conducted for each mutant.
RESULTS
Clinical characteristics
The cohort included 32 individuals, 12 female and 20 male. All participants underwent ES for developmental delay and/or congenital heart defects and were found to have a de novo variation in CHD4 except for one case clinically suspected to be inherited from an affected mother, but for whom a maternal sample was not available for testing. The age at last examination was between the newborn period and 30 years, with a mean and median age of 10 years. Figure 1b, Fig. 2, and Table 1S detail the clinical findings in the cohort. The majority of individuals were born after an uneventful pregnancy with an average birth weight and head circumference. Most participants were reported to have developmental delay. Speech delay was evident in 93% of participants (29/31), but absent speech was not reported. A cognitive assessment was performed in the majority of school-aged participants, which identified ID in the mild to moderate range in 86% (19/22). Three individuals (13%) had an IQ score in the normal range; one female had autism. Hypotonia was reported in 17/23 individuals (74%), and 26/31 had gross motor delays (83%). The average age of independent ambulation was 30 months. In three cases, independent ambulation was achieved after 5 years of age. Macrocephaly (head circumference above 95%ile) was frequent (39%, 11/28) but not a universal finding, and two individuals had microcephaly. Abnormal brain imaging was seen in 69% overall, and in 96% of the participants that underwent brain magnetic resonance imaging (MRI) (Fig. 2). A range of findings were reported including ventriculomegaly, hydrocephalus requiring shunting, Chiari 1, and a thin corpus callosum. Interestingly, two participants were diagnosed with moyamoya disease and subsequent stroke; one child had a congenital stroke and another presented during infancy. Congenital heart defects were present in 19/29 participants (65%). There was a variety of cardiac anomalies including septal defects, conotruncal anomalies, and valve anomalies (Fig. 2). Conductive and/or sensorineural hearing loss was present in 11/20 individuals (55%). Skeletal and limb anomalies, such as vertebral fusion and carpal/tarsal coalition, syndactyly and polydactyly, were present in 10 individuals (31%). Ophthalmic abnormalities such as strabismus, hypermetropia, and astigmatism were seen in 14/19 participants (73%). An anteriorly placed anus and vesicoureteral reflux were detected in three individuals. Additional various abnormalities were detected on physical examination or a skeletal survey, as described in Table 1S. Cryptorchidism and/or a microphallus were evident in 13 of the 20 males (65%), and low gonadotropin levels were documented in five of them. Hypogonadism was not detected in females; however, hormone levels were available for only two postpubertal females. Three individuals had documented growth hormone deficiency and one was treated with growth hormone supplements. Two individuals in the cohort died—one after birth due to complications of his congenital heart malformation and another at the age of 21 years due to complications of cervical vertebrae instability and long-term tracheostomy.

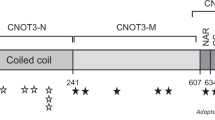
(a) CHD4 domains and the location of variants identified in this cohort. Each variant is marked by an asterisk (*). Dark gray asterisk represents a truncating variant at the marked position. (b) A table demonstrating the amino acid positions and the clinical findings of CHD4 variants detected in 32 individuals. The variants in bold have not been previously published. The domain color corresponds to (a). Clinical findings marked in gray represent patients with truncating variants. Hypogonadism refers to undescended testis, micropenis, or abnormal gonadotropin levels. (c) 3D protein modeling of the different CHD4 domains. The domain color corresponds to (a), and the variants are depicted in gold. Pathogenic variants were found in all highly conserved domains. The box shows that the Cys467 in the PHD2 domain is involved in zinc binding (purple sphere).

(a) The frequency of different clinical features in the CHD4-related syndrome cohort. The n refers to the number of individuals for whom we had data on the specific feature. For instance, only 23 individuals in the cohort had a brain magnetic resonance image (MRI) and 29 had an echocardiogram. (b) Histogram of types of brain and heart anomalies seen in individuals in this cohort.
Dysmorphology
Most participants were reported to have dysmorphic features. We reviewed the photographs of 26 participants (81%). Figure 3a includes photographs from 17 participants who consented for photo publication. Most, but not all, had a common facial gestalt that included a high and broad forehead, squared face, periorbital fullness, widely spaced eyes, short nose, and small or dysmorphic ears. These facial characteristics appear to be more evident during infancy and childhood. We used the image analysis based on deep convolutional neural network architecture provided by Face2Gene (FDNA Inc., USA)20 on 16 photographs of individuals with variants in the ATPase/helicase domain of CHD4 (Fig. 3b). The facial features of this cohort differed significantly from a control group of 32 healthy individuals (area under the curve [AUC] 0.917 of receiver operating characteristic [ROC] curve, p value 0.009).

(a) Photographs of 17 individuals with the CHD4-related syndrome at different ages. Photos 14, 19, and 23 were previously published in ref. 1 Photos 8 and 17 were published in ref. 2 (b) A composite of 16 photographs of participants harboring variants within the SNF2-like domain compared with healthy controls (FDNA Inc. USA).
Genotype–phenotype correlations
The majority of variants were missense substitutions or in-frame indels (91%) affecting the SNF2-like region of the protein (82%) (Fig. 1). Six individuals had missense substitutions in the N-terminal ATPase domain and 16 individuals in the C-terminal helicase domain. Six cases had missense variants in other conserved domains including the N-terminal CHDNT domain, the second PHD finger (PHD2), the second chromodomain, and in a conserved region in the C-terminal portion (DUF1087). The participants with variants in domains outside of the SNF2-like region had a phenotype and dysmorphologies similar to those of patients with variants within the SNF2-like region. There were three cases with predicted protein truncation (9%). For these individuals, the phenotype involved fewer systems and the facial features were not similar (Fig. 1b in gray, photos not shown). One individual with a c.2143C>T (p.Gln715*) variant had developmental delay, macrocephaly, and Pierre Robin sequence. Another individual with a c.2662delC (p.Val888Tyrfs*2) had developmental delay and mild ventriculomegaly. The individual with the C-terminal c.5608C>T (p.R1870*) variant had developmental delay, a thin corpus callosum, and another molecular and clinical diagnosis of FLNA-related periventricular nodular heterotopia.
Protein modeling
The predicted 3D structure of the different CHD4 domains is shown in Fig. 1c. All missense substitutions affect highly conserved residues (Fig. 1S, Table 2S). The majority of variants are located in the SNF2-like region (positions ~724–1281), especially its C-terminal portion. We could not identify a common denominator for the substitutions, as both exposed and buried amino acids were affected, only some of which were in a defined structural element such as an ɑ-helix. This may indicate both a structural and a functional effect. Only a few variants were located in other domains and, interestingly, some were in previously established functional positions: the p.Met202Ile variant is located in a region of the CHDNT domain that binds DNA and poly (ADP-ribose),21 the p.Cys467 position in the PHD2 zinc finger is predicted to participate in Zinc binding (Fig. 3c),22 and the p.Ser470 position is located within an H3-binding surface.22 The p.Arg1870* variant in the C-terminal end is predicted to result in a truncated protein lacking a highly conserved chain in the CHDCT2 domain (Fig. 1S).
ATPase and nucleosome remodeling studies
To understand the effect of CHD4 variants on protein function, we investigated the ATPase and nucleosome remodeling activity of five variants located in different protein domains: c.1400G>A (p.Cys467Tyr) in the PHD2 domain, c.1933C>T (p.Arg645Trp) in the chromodomain, c.2860A>G (p.Met954Val) in the N-terminal ATPase, c.3443G>T (p.Trp1148Leu) in the C-terminal helicase lobe, and c.4018C>T (p.Arg1340Cys) DUF1087 (Fig. 1). The variants in the PHD and chromodomains were found to impact ATPase activity and decrease it by 30–50% (Fig. 4). These variants also had decreased nucleosome remodeling activity (Fig. 5), which we hypothesize to be secondary to the decreased ATPase activity. The other variants had no impact on ATP hydrolysis. However, the p.Trp1148Leu and the p.Arg1340Cys variants had a significant decrease in the ability of CHD4 to remodel nucleosomes as demonstrated by a restriction enzyme accessibility assay on a nucleosome substrate (Fig. 5). The p.Met954Val in the N-terminal ATPase had a small increase in the remodeling activity, which was not statistically significant. In addition, we reviewed previous studies that evaluated ATPase hydrolysis and nucleosome remodeling activity of variants in CHD4 and its paralog CHD3.12,23 The majority of previously investigated variants had decreased ATPase hydrolysis and/or nucleosome remodeling activity. Increased remodeling was previously observed in a CHD3 variant in the N-terminal ATPase domain (p.Leu905Phe for the equivalent CHD4 position),12 and another CHD4 variant detected in endometrial carcinoma in the C-terminal helicase domain (p.His1196Tyr).23 A more detailed description of the functional data on specific variants from this study and previous ones is summarized in Table 2S.

Radiometric ATPase assays were performed in the presence of recombinant nucleosome, naked DNA, or in the absence of CHD4 (no protein). The percent of adenosine triphosphate (ATP) hydrolyzed was quantified for each mutant and then normalized to wild-type (WT) activity. Experimental data are presented as means with standard deviation. Individual data points are represented by black (nucleosome) or gray (naked DNA) squares. Three individual experiments from two individual purifications (n = 6) were performed. The significance of observed differences in activity was assessed by two-way analysis of variance (ANOVA) with Dunnett’s multiple test correction. Asterisk (*) indicates P values <0.05.

Chromatin remodeling assay. (a) Remodeling activity of CHD4 wild-type (WT) and mutants was assessed and quantified. 12, 6, and 3 nM enzyme was incubated with 12 nM recombinant nucleosome in the presence of HhaI. Digested DNA fragments were analyzed on 6% native polyacrylamide gel. (b) Quantitation of remodeling activity. Experimental data are presented as means with standard deviation. Three individual experiments from two individual purifications (N = 6) were performed. (c) A two-way analysis of variance (ANOVA) with Dunnett’s multiple test correction was performed to assess the significance of observed differences in activity. Table shows the significant P values for each variant and at each dose. Ns not significant.
DISCUSSION
The phenotype observed in individuals with SIHIWES involves multiple systems, similar to other Mendelian disorders affecting the epigenetic machinery.24,25 This is also supported by the wide expression of CHD4 during mammalian embryonic development, and its role in regulating gene expression and tissue differentiation in the brain, heart, and additional tissues.11,26,27,28,29,30,31,32 The majority of individuals in this cohort had global developmental delay, mild ID, dysmorphic features, and various congenital anomalies. However, there was variability in the extent and severity of clinical manifestations among different participants, which could not be clearly explained by the variant type or location. For example, the Arg1127Gln variant was seen in two participants; one of them had moderate ID and the other had a normal IQ. Similarly, there was no concordance in the degree of ID between the two patients with a variant in the Arg1183 position (Fig. 1b). The most common congenital anomalies affected the brain and the heart. Other common abnormalities included hypogonadism, limb and skeletal anomalies, conductive and or sensorineural hearing impairment, and various ophthalmic abnormalities. A few individuals in this cohort had hydrocephalus, Chiari 1, and cervical spine anomalies, and therefore, brain and cervical spine imaging is important in this condition. Other recommended evaluations include an echocardiogram, abdominal ultrasound, a skeletal survey, and audiologic and ophthalmologic evaluation. Interestingly, two participants had early-onset moyamoya disease and presented with congenital stroke or during infancy. Therefore, there should be a high level of suspicion for this diagnosis in SIHIWES patients. In terms of endocrine abnormalities, there was a high prevalence of hypogonadotrophic hypogonadism in males, and a few participants had low growth hormone levels. An endocrine assessment is recommended at the time of diagnosis and at puberty. The etiology of the hypogonadism is unknown, and it is not clear whether females with the condition may be at risk for hypogonadism as well. At this time, we know of normal postpubertal follicle-stimulating hormone (FSH) and luteinizing hormone (LH) levels in two affected females, one of whom was born to a mother who was suspected to be affected. The other females in this cohort are too young for assessment.
Most individuals in this cohort had a similar facial gestalt. However, the dysmorphic features were relatively minor and may be seen in other disorders. Generally speaking, this condition is not easily recognized in the clinical setting so it is likely to be diagnosed through a gene panel or ES. Still, the fact that SIHIWES is caused mainly by missense substitutions complicates variant interpretation, so in some cases the facial features and other specific clinical findings such as hypogonadism and bone fusions can aid in the interpretation of variants of uncertain significance found in CHD4.
Similarly to previous publications, the majority of CHD4 variants in this cohort were nontruncating. Most, but not all, variants were located within the SNF2-like region. The SNF2-like region contains the N-terminal ATPase domain and the C-terminal helicase domain, which belong to a larger family of proteins with similar enzymatic activity.8 In this cohort, six individuals had missense substitutions in the N-terminal ATPase domain and 15 individuals had missense substitutions in the C-terminal helicase domain. Almost 50% of these variants occurred in a hot-spot region between positions 1127 and 1192, which contain motifs V, Vb, and VI of SNF2.33 A severe and multisystemic presentation was observed in both domains (Fig. 1b). Interestingly, even for the recurrent variants in positions Arg1127, Arg1173, and Arg1183, there was variability in the degree of systemic involvement and cognitive impairment. As such, we did not identify genotype–phenotype correlations within this region.
ATPase hydrolysis and chromatin remodeling were previously shown to be disrupted in the presence of missense substitutions in the ATPase and helicase domains.12,23 Most previously investigated variants had decreased ATPase and remodeling activities, but a few variants had increased ATPase and remodeling activities or no effect at all.12,23 These properties do not appear to be related to a specific domain and are variant-specific (see Table 2S for details). In this study, we demonstrate a similar phenomenon. The p.Trp1148Leu variant within the helicase domain had decreased remodeling activity. However, the p.Met954Val variant in the ATPase domain was associated with normal ATPase activity (Fig. 4) and a small increase in nucleosome remodeling, which was not statistically significant (Fig. 5). It is not clear whether the small increase demonstrated in vitro is in fact significant in vivo, or whether there might be a different mechanism of pathogenicity. Since the mutated protein could be expressed and purified intact from human cells in culture, it is likely to be stable and normally expressed in vivo. The Met954 position is located in a highly conserved region, and the subject had a de novo variation and a systemic phenotype consistent with SIHIWES; therefore this is a likely pathogenic variant.
Six individuals with variants in the N-terminal CHDNT domain, the PHD2 zinc finger, the second chromodomain, and the C-terminal DUF1087 had a multisystemic phenotype and facial dysmorphologies consistent with SIHIWES, suggesting variants in these loci have a similar effect on CHD4 function. This was also supported by our functional assays performed on mutant CHD4 proteins expressing variants from the PHD2 zinc finger (p.Cys467Tyr), chromodomain (p.Arg645Trp), and DUF1087 (p.Arg1340Cys). The p.Cys476Tyr and p.Arg645Trp variants caused decreased ATP hydrolysis and nucleosome remodeling, and the p.Arg1340Cys variant led to decreased nucleosome remodeling activity. The present report for functional effects of variants in the second chromodomain and DUF1087 for CHD proteins is the first to the best of our knowledge. Our clinical observations and functional assays support the crucial role of these domains in CHD4 function during development. In addition, at the clinical level, there appears to be no difference in the phenotype observed in individuals with variants within or outside the SNF2-like ATPase/helicase domains. However, we only had six cases with nontruncating non-ATPase/helicase variants, and information from additional patients is required to establish the phenotypic spectrum of such individuals and possible genotype–phenotype correlations.
In this study we had three cases with truncating variants. The p.Arg1870* is predicted to result in a truncated protein that probably does not undergo nonsense-mediated decay (NMD).34 Therefore, for this variant a dominant negative mechanism is possible. The p.Gln715* and p.Val888fs variants are predicted to result in a protein lacking the entire or partial ATPase/helicase domains. Currently, blood or skin samples from these patients are not available, and we do not know whether the variants result in NMD or a truncated protein product. Although these variants occurred de novo, they may not be disease causing, especially considering their phenotype involves only a few systems and could result from a different syndrome not diagnosed through standard testing. Another possibility would be that truncating variants cause a milder phenotype.
A high prevalence of missense variants is also present in the chromatin modulation disorders CHD3-related intellectual disability and in the equivalent units of the SWI/SNF complex SMARCA4 and SMARCA2, which are associated with Coffin–Siris and Nicolaides–Baraitser syndromes, respectively.12,35 The similarity between the amino acid sequence and functional domains of these proteins and the presence of mainly missense variants in patients may suggest a common molecular mechanism. To date, there is accumulating data suggesting nontruncating variants in these genes have a dominant negative or gain-of-function effect. Specifically, for CHD4 and CHD3, the are no reports of small deletions that include these genes in individuals with developmental delays. The haploinsufficiency index is 9.9 and 16.9, respectively (https://decipher.sanger.ac.uk/); values above 10 are considered borderline and do not strongly support haploinsufficiency as the disease mechanism. According to the DECIPHER database, there are inherited cases of SMARCA2 deletions, suggesting SMARCA2 deletion does not cause Coffin–Siris syndrome. Truncating variants in SMARCA4 cause a cancer predisposition syndrome and not Nicolaides–Baraitser syndrome.36 Furthermore, Hodges et al. demonstrated that missense variants in SMARCA4, but not haploinsufficiency of SMARCA4, alter genome-wide DNA accessibility to enhancers.37 Another recent study showed that overexpression of dMi-2 mutants observed in malignancies disrupted Drosophila wing differentiation, also supporting a dominant negative mechanism.23 We speculate that nontruncating variants in CHD4 result in a mutant protein that assembles into the NuRD complex and binds nucleosomes, but has aberrant function that results in abnormal gene expression during development. Further research is required to fully understand the disease mechanism.
In conclusion, the CHD4-related syndrome is an autosomal dominant disorder characterized by developmental delay, varying degrees of cognitive disability, congenital anomalies, and dysmorphism. Based on a series of 32 patients, we could not establish genotype–phenotype correlation. Further research is required to better understand the disease mechanism and to determine the pathogenicity of truncating variants in this condition.
Change history
17 December 2019
In the original version of this Article the author Solveig Heide was incorrectly given as Heide Solveig. There was also an error in the spelling of the author Sandra Whalen, which was given as Sandra Wallen. In addition, the affiliation for Sandra Whalen was mistakenly quoted as AP-HP, Département de Génétique, Centre de Référence Maladies Rares “Anomalies du développement et syndromes malformatifs” Hôpital de la Pitié Salpêtrière, Paris, France instead of UF de génétique clinique, Centre de Référence Maladies Rares des Anomalies du développement et syndromes malformatifs, APHP, Hôpital Trousseau, Paris, France. All errors are now corrected in both the PDF and HTML versions of the Article.
17 December 2019
A Correction to this paper has been published: https://doi.org/10.1038/s41436-019-0727-3
References
Weiss K, Terhal PA, Cohen L, et al. De novo mutations in CHD4, an ATP-dependent chromatin remodeler gene, cause an intellectual disability syndrome with distinctive dysmorphisms. Am J Hum Genet. 2016;99:934–941.
Sifrim A, Hitz M-P, Wilsdon A, et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet. 2016;48:1060–1065.
Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer. 2011;11:588–596.
Xue Y, Wong J, Moreno GT, et al. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell. 1998;2:851–861.
Basta J, Rauchman M. The nucleosome remodeling and deacetylase complex in development and disease. Transl Res. 2015;165:36–47.
Woodage T, Basrai MA, Baxevanis AD, et al. Characterization of the CHD family of proteins. Proc Natl Acad Sci USA. 1997;94:11472–11477.
Wade PA, Jones PL, Vermaak D, Wolffe AP. A multiple subunit Mi-2 histone deacetylase from Xenopus laevis cofractionates with an associated Snf2 superfamily ATPase. Curr Biol. 1998;8:843–846.
Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 2011;21:396–420.
Clapier CR, Iwasa J, Cairns BR, Peterson CL. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat Rev Mol Cell Biol. 2017;18:407–422.
Hoffmeister H, Fuchs A, Erdel F, et al. CHD3 and CHD4 form distinct NuRD complexes with different yet overlapping functionality. Nucleic Acids Res. 2017;45:10534–10554.
Nitarska J, Smith JG, Sherlock WT, et al. A functional switch of NuRD chromatin remodeling complex subunits regulates mouse cortical development. Cell Rep. 2016;17:1683–1698.
Snijders Blok L, Rousseau J, Twist J, et al. CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language. Nat Commun. 2018;9:4619.
Thompson PM, Gotoh T, Kok M, et al. CHD5, a new member of the chromodomain gene family, is preferentially expressed in the nervous system. Oncogene. 2003;22:1002–1011.
Watson AA, Mahajan P, Mertens HDT, et al. The PHD and chromo domains regulate the atpase activity of the human chromatin remodeler CHD4. J Mol Biol. 2012;422:3–17.
Morra R, Lee BM, Shaw H, et al. Concerted action of the PHD, chromo and motor domains regulates the human chromatin remodelling ATPase CHD4. FEBS Lett. 2012;586:2513–2521.
Biasini M, Bienert S, Waterhouse A, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42(W1):1–7.
Yang J, Yan R, Roy A, et al. The I-TASSER suite: protein structure and function prediction. Nat Methods. 2014;12:7–8.
Taguchi H, Horikoshi N, Arimura Y, Kurumizaka H. A method for evaluating nucleosome stability with a protein-binding fluorescent dye. Methods. 2014;70:119–126.
Liu X, Li M, Xia X, et al. Mechanism of chromatin remodelling revealed by the Snf2-nucleosome structure. Nature. 2017;544:440–445.
Gurovich Y, Hanani Y, Bar O, et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat Med. 2019;25:60–64.
Silva AP, Ryan DP, Galanty Y, et al. The N-terminal region of chromodomain helicase DNA-binding protein 4 (CHD4) is essential for activity and contains a high mobility group (HMG) box-like-domain that can bind poly(ADP-ribose). J Biol Chem. 2016;291:924–938.
Mansfield RE, Musselman CA, Kwan AH, et al. Plant homeodomain (PHD) fingers of CHD4 are histone H3-binding modules with preference for unmodified H3K4. J Biol Chem. 2011;286:11779–11791.
Kovač K, Sauer A, Mačinković I, et al. Tumour-associated missense mutations in the dMi-2 ATPase alters nucleosome remodelling properties in a mutation-specific manner. Nat Commun. 2018;9:2112.
Bjornsson HT. The Mendelian disorders of the epigenetic machinery. Genome Res. 2015;25:1473–1481.
Larizza L, Finelli P. Developmental disorders with intellectual disability driven by chromatin dysregulation: clinical overlaps and molecular mechanisms. Clin Genet. 2019;95:231–240.
Sun F, Yang Q, Weng W, et al. Chd4 and associated proteins function as corepressors of Sox9 expression during BMP-2-induced chondrogenesis. J Bone Min Res. 2013;28:1950–1961.
Ingram KG, Curtis CD, Silasi-Mansat R, et al. The NuRD chromatin-remodeling enzyme CHD4 promotes embryonic vascular integrity by transcriptionally regulating extracellular matrix proteolysis. PLoS Genet. 2013;9:e1004031.
Sparmann A, Xie Y, Verhoeven E, et al. The chromodomain helicase Chd4 is required for Polycomb-mediated inhibition of astroglial differentiation. EMBO J. 2013;32:1598–1612.
Yamada T, Yang Y, Hemberg M, et al. Promoter decommissioning by the NuRD chromatin remodeling complex triggers synaptic connectivity in the mammalian brain. Neuron. 2014;83:122–134.
Walz K, Caratini-Rivera S, Bi W, et al. Modeling del(17)(p11.2p11.2) and dup(17)(p11.2p11.2) contiguous gene syndromes by chromosome engineering in mice: phenotypic consequences of gene dosage imbalance. Mol Cell Biol. 2003;23:3646–3655.
Wilczewski CM, Hepperla AJ, Shimbo T, et al. CHD4 and the NuRD complex directly control cardiac sarcomere formation. Proc Natl Acad Sci USA 2018;115:6727–6732.
Denner DR, Rauchman M. Mi-2/NuRD is required in renal progenitor cells during embryonic kidney development. Dev Biol. 2013;375:105–116.
Richmond E, Peterson CL. Functional analysis of the DNA-stimulated ATPase domain of yeast SW12/SNF2. Nucleic Acids Res. 1996;24:3685–3692.
Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci. 1998;23:198–199.
Bögershausen N, Wollnik B. Mutational landscapes and phenotypic spectrum of SWI/SNF-related intellectual disability disorders. Front Mol Neurosci. 2018;11:1–18.
Hasselblatt M, Nagel I, Oyen F, et al. SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol. 2014;128:453–456.
Hodges HC, Stanton BZ, Cermakova K, et al. Dominant-negative SMARCA4 missense mutations alter the accessibility landscape of tissue-unrestricted enhancers. Nat Struct Mol Biol. 2018;25:61–72.
Acknowledgements
We are grateful to the patients and their families for consenting to participate in this publication. This work was supported, in part, by the Intramural Research Program of the National Institute of Environmental Health Science, NIH (ES101965 to P.A.W.). The DDD study presents independent research commissioned by the Health Innovation Challenge Fund [grant number HICF-1009-003], a parallel funding partnership between Wellcome and the Department of Health, and the Wellcome Sanger Institute [grant number WT098051]. The views expressed in this publication are those of the author(s) and not necessarily those of Wellcome or the Department of Health. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). The Pediatric Cardiac Genomics Consortium (PCGC) program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, and U.S. Department of Health and Human Services through grants UM1HL128711, UM1HL098162, UM1HL098147, UM1HL098123, UM1HL128761, and U01HL131003. This manuscript was prepared in collaboration with investigators of the PCGC and has been reviewed and/or approved by the PCGC. PCGC investigators are listed at https://benchtobassinet.com/Centers/PCGCCenters.aspx. Some of the participants in the study were contacted through GeneDX (Gaithersburg, MD) and through GeneMatcher (https://www.genematcher.org/). The views expressed in this article reflect the results of research conducted by the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, or the United States Government. Dr. Weiss is supported by the Clinical Research Institute at Rambam.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Weiss, K., Lazar, H.P., Kurolap, A. et al. The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype–phenotype correlations, and molecular basis. Genet Med 22, 389–397 (2020). https://doi.org/10.1038/s41436-019-0612-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0612-0
Keywords
This article is cited by
-
Genetic and phenotypic analysis of 225 Chinese children with developmental delay and/or intellectual disability using whole-exome sequencing
BMC Genomics (2024)
-
Choroid plexus NKCC1 mediates cerebrospinal fluid clearance during mouse early postnatal development
Nature Communications (2021)
-
The emerging role of chromatin remodelers in neurodevelopmental disorders: a developmental perspective
Cellular and Molecular Life Sciences (2021)
-
Missense and truncating variants in CHD5 in a dominant neurodevelopmental disorder with intellectual disability, behavioral disturbances, and epilepsy
Human Genetics (2021)
-
A population-based approach for gene prioritization in understanding complex traits
Human Genetics (2020)
