
Abstract
Purpose
A new syndrome with hypotonia, intellectual disability, and eye abnormalities (HIDEA) was previously described in a large consanguineous family. Linkage analysis identified the recessive disease locus, and genome sequencing yielded three candidate genes with potentially pathogenic biallelic variants: transketolase (TKT), transmembrane prolyl 4-hydroxylase (P4HTM), and ubiquitin specific peptidase 4 (USP4). However, the causative gene remained elusive.
Methods
International collaboration and exome sequencing were used to identify new patients with HIDEA and biallelic, potentially pathogenic, P4HTM variants. Segregation analysis was performed using Sanger sequencing. P4H-TM wild-type and variant constructs without the transmembrane region were overexpressed in insect cells and analyzed using sodium dodecyl sulfate–polyacrylamide gel electrophoresis and western blot.
Results
Five different homozygous or compound heterozygous pathogenic P4HTM gene variants were identified in six new and six previously published patients presenting with HIDEA. Hypoventilation, obstructive and central sleep apnea, and dysautonomia were identified as novel features associated with the phenotype. Characterization of three of the P4H-TM variants demonstrated yielding insoluble protein products and, thus, loss-of-function.
Conclusions
Biallelic loss-of-function P4HTM variants were shown to cause HIDEA syndrome. Our findings enable diagnosis of the condition, and highlight the importance of assessing the need for noninvasive ventilatory support in patients.
Similar content being viewed by others


INTRODUCTION
Prolyl 4-hydroxylases (P4Hs) are key enzymes in the synthesis of collagens and the regulation of oxygen homeostasis.1,2,3 Previously, variants in the P4HA2 (OMIM 600608) encoding collagen prolyl 4-hydroxylase (C-P4H) ɑ-2 subunit have been associated with nonsyndromic high myopia (OMIM 617238),4 and variants in the P4HB (OMIM 176790) encoding prolyl 4-hydroxylase β-subunit have been associated with Cole–Carpenter syndrome (OMIM 112240),5 while variants in the EGLN1 (OMIM 606425) coding for a hypoxia-inducible factor (HIF) isoenzyme HIF-P4H/PHD2 have been associated with familial erythrocytosis and high altitude adaptation hemoglobin.6,7,8,9 A transmembrane prolyl 4-hydroxylase (P4H-TM) is encoded by the P4HTM gene (OMIM 614584), which has not yet been unequivocally linked to any Mendelian disorder. P4H-TM has its highest expression in the brain and eye,10,11 and it participates in the oxygen-dependent regulation of the hypoxia-inducible factor (HIF). The normal functioning of P4H-TM has been assessed to be crucial for normal vision and renal function in mice.12 However, the relevance of P4H-TM to human disease remains to be clarified.
In a previous study, we identified a large Finnish family with multiple affected family members presenting with a syndromic form of severe cognitive impairment.13 All the affected individuals had profound intellectual disability, hypotonia, and strabismus; we proposed the acronym HIDEA (hypotonia, intellectual disability, and eye abnormalities) for this condition. Our study identified a recessive disease locus in the chromosomal region 3p22.1-3p21.1. However, because all affected individuals carried potentially pathogenic biallelic variants in three genes, P4HTM, TKT, and USP4, segregating with the disease, the causative gene remained elusive.
In the current study, we report seven novel patients from four unrelated families presenting with HIDEA. DNA was available from six patients who carried potentially pathogenic biallelic P4HTM gene variants. One of the patients carried the same previously published homozygous potentially pathogenic variants in P4HTM, TKT, and USP4 genes. Five of the patients had novel homozygous or compound heterozygous variants only in the P4HTM gene, thereby indicating that P4HTM is the causative gene. Variant characterization demonstrated that the variants yield an insoluble protein product. In summary, we present robust evidence that HIDEA is caused by biallelic pathogenic P4HTM gene variants, and delineate the clinical characteristics of this syndrome.
MATERIALS AND METHODS
We conducted clinical and molecular genetic studies on seven patients from four families presenting with the phenotype resembling previously described patients with HIDEA syndrome.13 Patients were enrolled in the study from four centers (Clinic for Children and Adolescents, Oulu University Hospital, Oulu, Finland; Texas Children’s Hospital, Houston, TX; Kaiser Franz Josef Hospital with G.v. Preyer Children’s Hospital, Vienna, Austria; Department of Clinical Genetics, Amsterdam UMC, University of Amsterdam, Amsterdam, The Netherlands). Novel patients were recruited in the study based on the clinical phenotypes and biallelic potentially pathogenic P4HTM variants in patients resembling patients with HIDEA syndrome previously described by Kaasinen et al., which allowed clinicians and relatives of patients with HIDEA syndrome and biallelic potentially pathogenic P4HTM variants to contact the corresponding author (L.A.).13 Furthermore, we re-evaluated the patients of the previously published study.13 Written informed consent was obtained from all the parents or guardians of the patients participating in this study, including permission to publish photos. The study was approved by the ethics committees of the Northern Ostrobothnia Hospital District at the Oulu University Hospital and Baylor College (Houston, TX) (for family 2).
Molecular genetic studies
We extracted genomic DNA from peripheral blood or buccal swab samples of the probands, their affected siblings, and their parents using standard methods. We performed exome sequencing (ES) at the Broad Institute of MIT and Harvard (Cambridge, MA) for family 1; at the Baylor Genetics Laboratory (Houston, TX) for family 2; at the Neuromuscular Research Department, Medical University of Vienna, Austria, for family 3; and at the Department of Clinical Genetics, Amsterdam UMC, University of Amsterdam, Amsterdam, The Netherlands, for family 4. Segregation of P4HTM variants within the families was confirmed by Sanger sequencing. More details on ES analyses are provided in the Supplementary Note including detailed clinical data.
Characterization of P4H-TM variants
Fibroblasts and myoblasts from healthy control individuals were cultured, RNA was isolated, and reverse transcription polymerase chain reaction (RT-PCR) was performed to identify the transcript variants for P4H-TM, as described in the Supplementary Note.
P4H-TM variants were generated to the pVL1392–P4H-TM88–502 construct10 using routine PCR with appropriate primers (available on request). To generate the baculoviruses, variant constructs were transfected into Sf9 cells in the presence of flashBAC DNA with baculoFECTIN II transfection reagent (Oxford Expression Technologies) according to the flashBAC transfection protocol. Transfection of the wild-type construct has been described previously.10 Baculovirus stocks were amplified twice, and the amplified virus stocks were used to infect Sf9 cells grown as adherent culture on 10-cm tissue culture plates in complete TNM-FH insect cell media (Sigma-Aldrich). Cells were harvested 72 hours after infection, washed with phosphate buffer saline (PBS), and stored at -80 °C until use.
For lysis, the cells were thawed, resuspended, and homogenized in 5 ml lysis buffer containing 10 mM Tris-HCl pH 7.8, 100 mM glycine, 150 mM NaCl, 20 mM imidazol, 2 mM CaCl2, 20 µM FeSO4, 0,1% Triton X-100, and 1X Complete EDTA-free protease inhibitor (Roche). The insoluble and soluble fractions were separated with a 15-minute centrifugation at 20,000 × g at 4 °C. Cell pellets were washed twice with 5 ml lysis buffer and centrifuged again after each wash. The supernatants with soluble fractions were pooled together. Finally, the cell pellet was resuspended in 5 ml of lysis buffer, and sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) samples were prepared of the insoluble and soluble fractions. Two 10% SDS-PAGE gels were run parallel to each other and a threefold excess of the soluble sample versus the insoluble sample was loaded to take into account the increased volume from the washes. Further, one of the gels was stained with Coomassie, while the other was transferred overnight (4 °C, 30 V) to a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked, incubated for two hours (4 °C) with 1:10,000 dilution of rabbit anti-P4H-TM antibody,10 and for one hour (4 °C) with 1:5000 dilution of HRP-conjugated goat antirabbit antibody (Dako Denmark A/S, Glostrup, Denmark). Finally, the membrane was incubated for one minute with Pierce ECL Western Blotting Substrate (Thermo Scientific) and for three minutes with Amersham Hyperfilm ECL (GE Healthcare).
Analyses of skeletal muscle biopsy and autopsy samples
As a part of clinical diagnostics, skeletal muscle biopsy samples had previously been taken from 3 of 13 patients due to suggested neurometabolic disease. Histology and histochemistry on skeletal muscle samples (patients 1, 2, and 5) and autopsy tissue samples (patient 13) were performed using standard methods. Further, the activities of mitochondrial respiratory chain enzyme complexes were determined from skeletal muscle biopsy samples, as described previously.14
RESULTS
Clinical delineation of the patients with P4HTM variants
Clinical findings of patients 1–7 compared with an update of the previously described patients13 are compiled in Table 1. Eight patients are currently alive (current age between 3 and 55 years), and five are deceased (age at death between 7 months and 61 years). Nine patients are male, and four are female. All the patients have hypotonia (N = 13/13), and either intellectual disability (N = 12/13) or developmental delay (1/13, patient 4 who died at the age of 7 months). Furthermore, the common presenting features include strabismus (N = 11/13, 85%), epilepsy (N = 10/13, 77%), and central or obstructive sleep apnea (N = 8/13, 62%). Nine patients have a medical history of pneumonia or recurrent pneumonias (N = 9/13, 69%). Four patients have had respiratory distress and required respiratory support during pneumonias, and three patients died during pneumonia or other respiratory tract infections. Some patients also show dysautonomia, including constipation (N = 6/13, 46%), recurrent hypothermia or hyperthermia (N = 4/13, 31%), and reduced sweating (N = 2/13, 15%). Further, some patients have suspected recurrent parasomnias (N = 4/13, 31%). Six patients (N = 6/13, 46%) have a body mass index (BMI) over 25 kg/m2. Two patients (N = 2/4, 50%) had advanced bone age at the age of 8 years. Brain magnetic resonance image (MRI) is normal in most patients.
Detailed clinical descriptions of the patients are provided in the Supplementary Note. The characteristic gait of patients 2, 3, 8, 10, and 11 is demonstrated in Supplementary Videos 1-5.
Histopathological and biochemical findings of skeletal muscle samples and brain autopsy sample
The muscle biopsy of patient 1 showed low activity of mitochondrial respiratory chain complex III. In addition, the muscle biopsy of patient 2 was suggestive of a mitochondrial, neurometabolic disease. Electron microscopy indicated increased mitochondria, with some that were enlarged and abnormally shaped. Mitochondrial respiratory chain activities revealed a decrease in complexes I–IV. Furthermore, the muscle biopsy of patient 5 showed an increased variability of muscle fiber diameters, hypertrophied muscle fibers, scattered small atrophied fibers, and basophilic muscle fibers (Supplementary Fig. 1). The muscle biopsy sample of patient 13, taken during autopsy, showed unspecific type 2 muscle fiber atrophy (Supplementary Fig. 2A) and COX-negative fibers suggestive of mitochondrial pathology. In addition to these findings in skeletal muscle samples, two patients (patients 1 and 13) had mild fatty degeneration of liver. Patient 13’s autopsy also revealed that the cerebellum was atrophic, and histologically there was both loss and degeneration of Purkinje cells (Supplementary Fig. 2B).
Analysis of the P4HTM gene
All seven Finnish patients (family 1, patient 1 and family 5, patients 8–13) had a homozygous P4HTM gene variant c.1073G>A (NM_177938.2, GRCh38 g.3:49005046G>A, rs182812551). While this variant is predicted to cause a missense change in the canonical transcript, which was not expressed in fibroblasts and myoblasts, it creates an in-frame loss of complete exon 6 of the main transcript (NM_177939.2) on the RNA level (r.888_1073del).13 The parents of the patients were heterozygous carriers of the variant. The variant is present in the Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/variant/3-49042479-G-A) database with a minor allele frequency (MAF) of 9.75 × 10−5; the allele frequency is 20× higher in Finland (7.8 × 10−4) than in non-Finnish Europeans (4 × 10−5), and there are no homozygous individuals.
Two brothers of family 2, patients 2 and 3, were compound heterozygous for the P4HTM gene c.482A>C; p.(His161Pro) (NM_177938.2, GRCh38 g.3:49001483A>C) and c.286dupC; p.(Gln96Profs*29) (NM_177938.2, GRCh38 g.3:48990542dupC) variants. The father was a heterozygous carrier of the P4HTM gene c.286dupC; p.(Gln96Profs*29) variant and the mother was a heterozygous carrier of the P4HTM gene c.482A>C; p.(His161Pro) variant. The healthy sister was heterozygous for the P4HTM gene c.482A>C; p.(His161Pro) variant and negative for the c.286dupC; p.(Gln96Profs*29) variant.
Two brothers of family 3, patients 5 and 6, were homozygous for the c.1594C>T; p.(Gln532*) (NM_177938.2, GRCh38 g.3:49006809C>T) variant. Their parents were heterozygous carriers. No DNA was available from patient 4, who presented with a similar clinical phenotype as his affected siblings.
Trio ES analysis of patient 7 (family 4) and his parents revealed that the patient had a homozygous variant in the P4HTM gene c.949delG (NM_177938.2, GRCh38 g.3:49004922delG). The parents carried this variant in a heterozygous state. This deletion predicts a frameshift change in the protein. Valine at position 317 is the first affected amino acid, with a premature stop codon after 28 amino acids p.(Val317Phefs*30). Frameshift variants creating a premature stop codon yield a shorter protein or no protein at all.
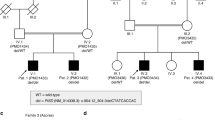
The pedigrees, identified variants, and their location in the protein are illustrated in Fig. 1. The genetic findings are described in Supplementary Table 1.

Pedigrees and Sanger validation and segregation of the identified P4HTM variants. (a) The segregation of the P4HTM c.1073G>A variant (NM_177938.2) in family 1. The protein defect of the biologically active main transcript is given in parenthesis (NM_177939.2). (b) The segregation of the compound heterozygous P4HTM c.286dupC; p.(Gln96Profs*29) and c.482A>C; p.(His161Pro) variants (NM_177938.2) in family 2. (c) The segregation of the P4HTM c.1594C>T; p.(Gln532*) variant (NM_177938.2) in family 3. (d) The segregation of the P4HTM c.949delG; p.(Val317Phefs*30) variant (NM_177938.2) in family 4. (e) Facial figure of patient 1 (F1:II.3) shows facial hypotonia with tented upper lip vermilion and open mouth posture, a round face, and strabismus. (f) Facial figure of patient 2 (F2:II.1) shows hypotonic/myopathic facial appearance with a tented upper lip vermilion and open mouth posture. (g) Facial figure of patient 3 (F2:II.3) shows marked facial hypotonia with open mouth posture, giving a myopathic facial appearance. (h) Depiction of the P4HTM protein and location of the variants identified in the four families.
Characterization of the variants
Previously, we characterized in vitro recombinant P4H-TM88–502 of the 502-residue noncanonical isoform, which lacks the transmembrane domain expressed in insect cells.10 Our RT-PCR analyses suggested that the 502-residue variant is the major transcript expressed from human-derived cells (Supplementary Fig. 3); therefore, we used it here to separately generate the variants corresponding to p.His161Pro, p.Gln532* and p.Arg296Ser;Val297_Arg358del on this backbone and expressed these variants in insect cells along with wild-type P4H-TM88-502. All variants yielded a recombinant enzyme analyzed using SDS-PAGE and Coomassie blue or western blotting (Fig. 2). However, the fraction of soluble protein was severely reduced for the p.Gln532* variant and completely abolished for the p.His161Pro and p.Arg296Ser;Val297_Arg358del variants (Fig. 2). Despite the lack of the C-terminal endoplasmic reticulum retention signal Arg-Val-Glu-Leu in the p.Gln532* variant, we were unable to detect it in the expression medium at the end of the 72-hour expression period (data not shown). The P4HTM variants studied here reside in different parts of the polypeptide and represent different types of variants. Despite these differences, the outcome is very similar. Although no structural data is available on P4H-TM, the insolubility of the variants suggests improper folding of the proteins. Together, these data suggest that the variants identified in the patients cause loss of function of P4H-TM.

Cells were lysed and the proteins were separated into insoluble (IS) and soluble (S) fractions. Both fractions of WT and variant enzymes p.His161Pro, p.Gln532*, and p.Arg296Ser;Val297_Arg358del (Ex6del) were loaded on 10% SDS-PAGE, and the gels were either stained with Coomassie blue (top) or western blotting against P4H-TM (bottom).
DISCUSSION
In this study, we report seven new patients from four unrelated families and a clinical update of a large previously published consanguineous family with HIDEA syndrome caused by biallelic P4HTM variants. The phenotype is characterized by muscular and central hypotonia, hypoventilation, intellectual disability, dysautonomia, epilepsy, and eye abnormalities (HIDEA). Variant characterization demonstrates that the variants affect protein folding by yielding an insoluble protein product.
P4H-TM has a broad expression pattern, with the highest expression levels in the brain and eye. All the patients with P4HTM gene variants have developmental delay or intellectual disability and visual abnormalities—such as strabismus, rotating eye movements, nystagmus, or cortical blindness—thereby suggesting that P4H-TM is crucial for the functioning of the normal human brain as well as the visual tract. Muscle biopsy samples from four patients demonstrate increased variability of the muscle fiber diameters, mild type 2 muscle fiber atrophy and COX-negative fibers, and decreased mitochondrial respiratory chain enzyme activities in two patients, thereby suggesting that a defect in P4H-TM may also disrupt normal mitochondrial function. The autopsy of a 61-year-old patient with the homozygous c.1073G>A variant revealed loss and degeneration of Purkinje cells, which could be caused by a long-term undiagnosed hypoventilation and/or neurodegeneration based on progressive neurodegenerative diseases.
P4H-TM knockout mice reveal overlapping phenotypic features with HIDEA patients with biallelic P4HTM variants, such as retinal defects causing vision impairment,12 thereby suggesting that loss-of-function is the possible underlying mechanism. Further, mouse studies suggest that P4H-TM is necessary for normal visual and renal function.12 As mentioned above, the patients exhibit various visual abnormalities, such as congenital nystagmus, strabismus, rotating eye movements, achromic retinal fundi, and cortical blindness. However, patients with P4HTM gene variants did not show any evidence of structural kidney abnormalities or albuminuria indicative of decreased kidney function, even at an older age.
In addition to the previously described combination of symptoms, this study reveals hypoventilation, including obstructive and central sleep apnea, dysautonomia, and an increased tendency to develop respiratory distress during pneumonias as novel characteristic clinical features of HIDEA patients. Several patients have BiPAP dependency at night due to central and obstructive sleep apnea or hypoventilation especially during respiratory infections requiring respiratory support. Four patients (4/13, 31%) had infantile onset tendency to respiratory insufficiency and respiratory arrest resulting in infantile or childhood death of three of them. There is heterogeneity in the clinical presentation of patients even with exactly the same pathogenic biallelic P4HTM variants suggesting that the disease severity is modified by additional genetic and environmental factors.
Further, we found that the P4HTM c.1073G>A variant creates an in-frame loss of exon 6 from the messenger RNA (mRNA) of the 502-residue transcript variant 1 (NM_177939.2), which yields an insoluble protein product. This is consistent with the latest research findings that pathogenic variants altering protein code often disrupt splicing.15 Interestingly, the noncanonical transcript (NM_177939.2) coding for the 502-residue protein was the prevalent transcript variant expressed in normal human fibro- and myoblasts instead of the canonical transcript (NM_177938.2) yielding a 563-residue protein. Theoretically, the canonical transcript coding for the 563-residue protein could be expressed in brain tissue, which is critical for HIDEA syndrome, but we do not have any experimental evidence to prove this. In northern blot analysis in brain the major P4HTM mRNA species was 2.3 kb, unlike in all other tissue studied, where it was 1.8 kb,10 which may indicate that in the brain the canonical transcript is expressed. Both transcripts have nine exons, but the transcript coding for the 502-residue protein excludes parts of exons 6 and 7. All the pathogenic variants identified in this study located in areas of the P4HTM sequence present in both transcripts, and it is currently unknown whether HIDEA syndrome could be caused by biallelic pathogenic variants located in parts of exons 6 or 7 that are not present in the transcript coding for the 502-residue protein.
P4H-TM was originally suggested to be a fourth HIF-P4H,16 although it was later reported to be more closely homologous to the C-P4Hs.10 Unlike neither of the previously characterized P4Hs, it has an N-terminal EF-hand motif16 that is implicated in calcium binding. Recombinant P4H-TM did not hydroxylate in vitro type I collagen polypeptide but it hydroxylated HIF1α.10 Its downregulation by small interfering (siRNA) stabilized HIFα,10 and its genetic knockout in mice associated with induction of renal erythropoietin expression similarly to inhibition of HIF-P4Hs.17 In cortical neurons genetic deficiency of P4H-TM stabilized HIF1α under normoxia, and in tissues with high endogenous P4H-TM expression level, such as brain, eye, and kidney, the expression of certain HIF target genes was more upregulated in hypoxia compared with wild-type mice.12 However, its location having the catalytic domain within the endoplasmic reticulum lumen, and its ability to also hydroxylate to a low extent HIFα polypeptide, where the HIF-P4H prolines were mutated to alanines, would suggest it may have other substrates than HIFα.10 The only known genetic variants of any HIF-P4H are those for HIF-P4H-2, the most abundant isoenzyme whose inhibition alone stabilizes HIFα3 that associate with erythropoiesis. As these variants did not cause a complete loss-of-function, and some even had a gain-of-function phenotype, it may be that complete loss of HIF-P4H-2 catalytic activity, which would result in a full-powered HIF response, is not tolerable in humans, similarly to mice.18 Therefore direct comparison of the phenotype of the loss-of-function P4HTM variants characterized here to regulation of the hypoxia response is not feasible.
Autosomal recessive inheritance is estimated to have a small contribution in the etiology of intellectual disability in outbred populations.19 Northern Finland is a known genetic isolate, where the current population has arisen from a small number of founder families, and enrichment of rare recessive gene variants has been demonstrated in this young population.20 All the Finnish patients in this study had the same variant in the P4HTM, thereby suggesting a founder effect.21 Homogeneous populations, such as the Finnish population, that share long segments of linkage disequilibrium are useful in mapping the causative locus of a monogenic disease; however, the homogeneous genetic background hinders the identification of the actual disease-causing gene as multiple variants in different genes in the same locus may cosegregate with the disease. In contrast, heterogeneous populations with marked genetic variability facilitate in underpinning the causative gene and broaden the knowledge of the full phenotypic spectrum caused by a specific gene. Studying patients with the same monogenic syndrome from different ethnic backgrounds has repeatedly revealed novel disease-causing genes. International collaborations may enable the identification of the causative gene and the delineation of the phenotype even in very rare childhood heritable diseases.22
In conclusion, we report seven new patients with the HIDEA syndrome and demonstrate that the phenotype in this cohort of patients is strikingly similar to the previously studied patients with the syndrome. Genetic analysis identified recessively inherited intragenic P4HTM variants as an underlying cause of the syndrome. Variant characterization suggested that the identified variants cause loss of function of P4H-TM. Novel, previously undescribed features include hypoventilation (most pronounced during respiratory infections), central and obstructive sleep apnea, and dysautonomia. This indicates the importance of performing polysomnography in patients with biallelic pathogenic P4HTM variants and assessing the possible need for noninvasive ventilatory support to prevent sudden death. Our results demonstrate that biallelic loss-of-function P4HTM gene variants cause HIDEA, and greatly facilitate the diagnosis and management of this challenging condition.
References
Myllyharju J. Prolyl 4-hydroxylases, key enzymes in the synthesis of collagens and regulation of the response to hypoxia, and their roles as treatment targets. Ann Med. 2008;40:402–417.
Myllyharju J. Prolyl 4-hydroxylases, master regulators of the hypoxia response. Acta Physiol (Oxf.). 2013;208:148–165.
Kaelin WG, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402.
Guo H, Tong P, Liu Y, et al. Mutations of P4HA2 encoding prolyl 4-hydroxylase 2 are associated with nonsyndromic high myopia. Genet Med. 2015;17:300–306.
Rauch F, Fahiminiya S, Majewski J, et al. Cole-Carpenter syndrome is caused by a heterozygous missense mutation in P4HB. Am J Hum Genet. 2015;96:425–431.
Percy MJ, Zhao Q, Flores A, et al. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc Natl Acad Sci USA 2006;103:654–659.
Percy MJ, Furlow PW, Beer PA, et al. A novel erythrocytosis-associated PHD2 mutation suggests the location of a HIF binding groove. Blood. 2007;110:2193–2196.
Ladroue C, Carcenac R, Leporrier M, et al. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med. 2008;359:2685–2692.
Lorenzo FR, Huff C, Myllymäki M, et al. A genetic mechanism for Tibetan high-altitude adaptation. Nat Genet. 2014;46:951–956.
Koivunen P, Tiainen P, Hyvärinen J, et al. An endoplasmic reticulum transmembrane prolyl 4-hydroxylase is induced by hypoxia and acts on hypoxia-inducible factor alpha. J Biol Chem. 2007;282:30544–30552.
Hyvärinen J, Parikka M, Sormunen R, et al. Deficiency of a transmembrane prolyl 4-hydroxylase in the zebrafish leads to basement membrane defects and compromised kidney function. J Biol Chem. 2010;285:42023–42032.
Leinonen H, Rossi M, Salo AM, et al. Lack of P4H-TM in mice results in age-related retinal and renal alterations. Hum Mol Genet. 2016;25:3810–3823.
Kaasinen E, Rahikkala E, Koivunen P, et al. Clinical characterization, genetic mapping and whole-genome sequence analysis of a novel autosomal recessive intellectual disability syndrome. Eur J Med Genet. 2014;57:543–551.
Hautakangas M-R, Hinttala R, Rantala H, et al. Evaluating clinical mitochondrial respiratory chain enzymes from biopsy specimens presenting skewed probability distribution of activity data. Mitochondrion. 2016;29:53–58.
Soemedi R, Cygan KJ, Rhine CL, et al. Pathogenic variants that alter protein code often disrupt splicing. Nat Genet. 2017;49:848–855.
Oehme F, Ellinghaus P, Kolkhof P, et al. Overexpression of PH-4, a novel putative proline 4-hydroxylase, modulates activity of hypoxia-inducible transcription factors. Biochem Biophys Res Commun. 2002;296:343–349.
Laitala A, Aro E, Walkinshaw G, et al. Transmembrane prolyl 4-hydroxylase is a fourth prolyl 4-hydroxylase regulating EPO production and erythropoiesis. Blood. 2012;120:3336–3344.
Takeda K, Ho VC, Takeda H, et al. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. 2006;26:8336–8346.
Rauch A, Wieczorek D, Graf E, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682.
Lim ET, Würtz P, Havulinna AS, et al. Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet. 2014;10:e1004494.
Norio R, Finnish Disease, Heritage I. characteristics, causes, background. Hum Genet. 2003;112:441–456.
Boycott KM, Rath A, Chong JX, et al. International cooperation to enable the diagnosis of all rare genetic diseases. Am J Hum Genet. 2017;100:695–705.
Acknowledgements
We thank all the families that participated in this study. Further, we acknowledge Pirjo Keränen, BCO sequencing core, for excellent technical assistance. This study was supported by the Academy of Finland Grants 266719 and 308009, the S. Jusélius Foundation, the Emil Aaltonen Foundation, and the Jane and Aatos Erkko Foundation to P.K. We also thank the Scribendi proofreading team for language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
X.W. and Y.Y.: The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular genetic testing offered at Baylor Genetics. The other authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Shared last author: Peppi Koivunen and Johanna Uusimaa
Shared second last author: Silvana van Koningsbruggen, Xia Wang and Wolfgang M. Schmidt
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. If you remix, transform, or build upon this article or a part thereof, you must distribute your contributions under the same license as the original. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/.
About this article

Cite this article
Rahikkala, E., Myllykoski, M., Hinttala, R. et al. Biallelic loss-of-function P4HTM gene variants cause hypotonia, hypoventilation, intellectual disability, dysautonomia, epilepsy, and eye abnormalities (HIDEA syndrome). Genet Med 21, 2355–2363 (2019). https://doi.org/10.1038/s41436-019-0503-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0503-4
Keywords
This article is cited by
-
Metabolic characteristics of transmembrane prolyl 4-hydroxylase (P4H-TM) deficient mice
Pflügers Archiv - European Journal of Physiology (2024)
-
Rare variant analyses across multiethnic cohorts identify novel genes for refractive error
Communications Biology (2023)
-
Biallelic P4HTM variants associated with HIDEA syndrome and mitochondrial respiratory chain complex I deficiency
European Journal of Human Genetics (2021)
