
Abstract
Purpose
Despite the successful progress next-generation sequencing technologies has achieved in diagnosing the genetic cause of rare Mendelian diseases, the current diagnostic rate is still far from satisfactory because of heterogeneity, imprecision, and noise in disease phenotype descriptions and insufficient utilization of expert knowledge in clinical genetics. To overcome these difficulties, we present a novel method called Xrare for the prioritization of causative gene variants in rare disease diagnosis.
Methods
We propose a new phenotype similarity scoring method called Emission-Reception Information Content (ERIC), which is highly tolerant of noise and imprecision in clinical phenotypes. We utilize medical genetic domain knowledge by designing genetic features implementing American College of Medical Genetics and Genomics (ACMG) guidelines.
Results
ERIC score ranked consistently higher for disease genes than other phenotypic similarity scores in the presence of imprecise and noisy phenotypes. Extensive simulations and real clinical data demonstrated that Xrare outperforms existing alternative methods by 10–40% at various genetic diagnosis scenarios.
Conclusion
The Xrare model is learned from a large database of clinical variants, and derives its strength from the tight integration of medical genetics features and phenotypic features similarity scores. Xrare provides the clinical community with a robust and powerful tool for variant prioritization.
Similar content being viewed by others

INTRODUCTION
Application of next-generation sequencing technologies have brought great progress in diagnosing the genetic cause of rare Mendelian diseases. More than 100 novel disease–gene associations were identified per year from 2012 to 2016 on average.1 However, the current diagnostic rate, which ranges from ~28% in exome sequencing2 to 57% in the most comprehensive family trio genome sequencing3 studies, is still far from satisfactory, and there are still more than 3000 (~50%) known OMIM diseases with unknown genetic causes.4 Thus prioritizing sequence variants explaining the disease phenotypes becomes crucial for genetic diagnosis of rare Mendelian disorders.
Several strategies have been developed to prioritize the pathogenic variants associated with rare disorders. One group of methods (e.g., MutationTaster,5 CADD,6 M-CAP,7 REVEL8) use genotype-only information (sequence and genomic attributes) to provide in silico prediction of variant pathogenicity. However, because each healthy person generally harbors about 100 loss-of-function deleterious variants,9 further consideration of genotype–phenotype association is needed for clinical applications. To further prioritize the variants, phenotype-driven methods (e.g., eXtasy,10 Exomiser,11 Phen-Gen12) had been proposed to combine the results of existing in silico prediction algorithms and a phenotypic relatedness measure, for the scoring and ranking of disease causative gene variants. However, even though these phenotype-driven methods have gained wide applications in clinical diagnosis, the diagnostic rate in real clinical settings is unsatisfactory and very far from the numbers shown in simulation studies.3 One potential reason for the discrepancy could be the incompleteness, heterogeneity, imprecision, and noise in disease phenotype descriptions. To overcome these challenges, we developed a new robust phenotype similarity score and a machine learning method (Xrare) jointly modeling phenotypic features and multiple genetic features including ACMG/AMP guideline-based features. ACMG/AMP guidelines are standards and guidelines for the interpretation of sequence variants released by the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP).13 The use of genetic features derived from ACMG/AMP guidelines allows our model to capture domain expert knowledge reflecting the best practice in medical genetics. For phenotypic features, the challenge is in ensuring that the features are tolerant of incompleteness (partial presentation of symptoms), imprecision (presenting symptoms less specific than the ones associated with the disease), and noise (presenting symptoms unrelated to the disease). Two recent exome sequencing studies14,15 with individual patient Human Phenotype Ontology (HPO) phenotypes demonstrated that 48% of patients had some phenotype noise and 25% patients had more than 50% noise if measured by HPO gene–phenotype exact associations (Supplementary Table S1). This highlighted the widespread existence of imprecise and noisy phenotypes in clinical settings. To handle these difficulties, we developed a new phenotype score called Emission-Reception Information Content (ERIC). ERIC can robustly measure the phenotypic similarity between imprecise and noisy patient phenotypes and known phenotypes associated with a disease or a gene. Through extensive simulations of spike-in synthetic genomes with various phenotype noise levels and real clinical data sets, we evaluated the variant prioritization performance of Xrare in comparison with a wide range of currently popular genotype-only and phenotype-driven methods and demonstrated the improvement from our method in rare disease diagnosis.
MATERIALS AND METHODS
Xrare model features
Xrare is a machine learning approach to disease-causing variant prioritization based on a rich set of phenotypic and genetic features. The full description of features in our predictive model is described in Supplementary Methods and summarized in Supplementary Table S2. Briefly, there are 51 features, including 6 population allele frequency–related features, 5 gene-phenotype similarity scores, 15 ACMG/AMP guideline-based features, 9 gene-level constraint scores, 12 existing in silico prediction scores of pathogenicity, 2 functional impact features of variants, and 2 database-related gene-level features. In particular, ACMG/AMP features reflect the current best practice in assessing pathogenicity of genetic variants by combining multiple categories of evidence. ERIC-based features, on the other hand, enable usage of phenotypic information not only in the case when the target gene has phenotypic annotations but also in the case when such annotations are not available. In the latter case, we obtain “predicted” phenotype similarity scores based on genes with phenotypic annotations and related to the target gene in terms of sequence similarity, pathway comembership, and other forms of interactome data (Supplementary Table S3).
Xrare model training
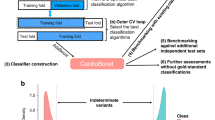
The schematic overview of the Xrare model is shown in Fig. 1a. We used 49,021 known pathogenic variants from ClinVar to train and validate our Xrare model (Supplementary Table S4). First, 41,590 variants identified by year 2011 were used to derive ACMG/AMP guideline–based features, because effectively implementing some guideline-based features (such as PS1, PM1, PM5, PP2) requires a large number of known pathogenic variants. Next, a gradient boosting decision tree (GBDT16) algorithm implemented in XGBoost17 was applied to learn the Xrare model based on training data derived from 6576 ClinVar variants identified in 2012–2015. GBDT, unlike the linear model, is robust to multicollinearity when features are redundant and highly correlated. More details in the construction of training data and model implementation are described in Supplementary Methods. Finally, 855 ClinVar variants identified since 2016 were used to evaluate performances. We also calculated the importance of all the features using the “xgb.importance” function in XGBoost, as shown in Fig. 1b and Supplementary Figure S1.

Schematic overview of the Xrare model and most important features for predicting variant pathogenicity. a Schematic overview of the Xrare model. The collected 49,021 pathogenic ClinVar variants were divided into three parts in terms of their publishing years: 41,590 variants identified by 2011 used for implementing American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) guideline evidence; 6576 variants spiked in synthetic genomes for model training; the remaining 855 variants identified since 2016 used for model evaluation. The preexisting in silico computation scores of variants, population-level scores, and other ACMG evidence scores were used as features for machine learning. Phenotype-related features came from gene–phenotype similarity Emission-Reception Information Content (ERIC) score and predicted gene–phenotype associations. b The top 15 most important features from Xrare model. See Supplementary Figure S1 for all features.
To prevent “data leakage” (double usage of information in both the evaluation set and model training) and model overfitting, we applied a series of strategies including separating spike-in pathogenic variants by years (Supplementary Table S4); distinct background genomes in feature calculation, model training, and evaluation; choosing only in silico prediction scores without strong publication year bias (Supplementary Figure S2); and carefully excluding known HPO phenotype associated genes before 2016 for novel gene performance evaluations (details in “Discussion”).
Integration of gene–phenotype annotation from human, mouse, and zebrafish
Measures of the similarity between two sets of phenotypes (e.g., between the phenotypes annotated to a gene and the phenotypes associated with a disease) usually depend on a phenotype ontology, which is composed of a set of phenotype terms hierarchically organized from general to specific phenotype descriptions. In this paper we used the HPO18 to encode phenotypes. Human phenotype annotations were downloaded from the official HPO website. We also downloaded zebrafish and mouse gene–phenotype annotations based on Uberpheno ontology19 (UPO), which is a cross-species phenotype ontology including mixed phenotype descriptions from human, mouse, and zebrafish. We converted any UPO term not in HPO into an HPO term based on the UPO structure (i.e., a non-HPO term was replaced by its ancestral HPO term with the greatest information content). In this way, 183,558 HPO terms were annotated to 8643 human genes.
ERIC is a new measure of similarity between phenotype terms
The poor performance of existing phenotype similarity measures in the presence of imprecision and noise motivated us to develop ERIC. The information content (IC) of a phenotype term t is computed as:
where N denotes the total number of genes under consideration, and n denotes the number of genes with phenotype t. The ERIC similarity between t1 and t2 is calculated as:
where tMICA is the most informative common ancestor (MICA) of t1 and t2. In addition, the ERIC score is set to have a minimum value of zero.
Here we briefly describe the rationale for the ERIC score formula. First, we define the distance between term t and its ancestral term tancestor as:
Then we use this formula to calculate how much IC emitted from t1 is received by t2:
In addition, to make the score symmetric between two phenotype terms, we replaced IC(t2) with the minimum of IC(t1) and IC(t2) in this formula. In addition, the ERIC score is set to have a minimum value of zero, when the minimum Dist (t,tMICA) of t1 and t2 is larger than IC(tMICA). Intuitively, this at tMICA with a radius of IC(tMICA) are considered noise. The final definition of ERIC similarity can be written as:
Other IC-based measures tested in the study, such as Resnik,20 Lin,21 and Jiang-Conrath22 (JC) measures, are calculated as:
Similarity between phenotype sets
Best match sum23 (BMS) was used to calculate the similarity between two sets of phenotype terms, the query phenotype set T1 and the annotation phenotype set T2:
More details are described in Supplementary Methods.
Gene–phenotype associations predicted over interactomes
Because only a subset of genes have phenotype annotations from HPO or UPO, we obtained phenotypic annotations for the remaining genes utilizing ten interactomes to capture different types of gene–gene interactions including information from Gene Ontology (GO) terms, protein domains, gene expression patterns, curated functional networks, and sequence similarities (Supplementary Table S3). First, for each of the ten interactomes, we obtained a predictor vector defined as the weighted phenotype similarity score for each gene–phenotype pair, using a subset of genes with phenotype annotations as seed genes. Then GBDT method was used to model the gene-phenotype score as predicted by the ten weighted phenotype similarity score vectors. After the model was trained, we obtained the predicted gene-phenotype similarity scores (Pred_phen score in Supplementary Table S2) for all genes. Details are described in Supplementary Methods.
Performance evaluation on variants in ClinVar since 2016
We synthesized 3420 patient genomes by inserting each of the 855 pathogenic variants identified since 2016 into 4 background genomes randomly selected from 400 healthy genomes (internal 30× genome sequencing data from people more than 50 years old and free of rare Mendelian disorders). We set the genotype of the spike-in variant to heterozygote if the variant is from a dominant disease gene, and homozygote if the variant is from a recessive disease gene. Among the 855 pathogenic variants, 417 variants are from “novel” genes that are not found to be associated with rare human diseases in literature before 2016. To evaluate the performance in different phenotype situations, phenotypes of a patient with a pathogenic variant of a gene known to be linked to rare diseases before 2016 were simulated by mixing different levels of precise, imprecise, and noisy phenotypes. In total, we created six phenotype simulation models: 1|2|0, 1|2|2, 2|2|0, 2|2|2, 3|2|0, 3|2|2, where x|y|z means x precise, y imprecise, and z noisy phenotypes for a simulated patient. Each of the synthesized genomes randomly chose one phenotype simulation model to use (see more simulation details in Supplementary Methods). HPO phenotypes of novel genes not linked to rare diseases before 2016 were manually curated in terms of the clinical descriptions from the OMIM website, which were used to evaluate the performance on novel genes.
Performance evaluation in real clinical data set
We also evaluated the performance of our method using two recently published real clinical data sets with HPO phenotypes and causal variants provided by clinicians. The first data set consists of 45 clinically confirmed variants from a study reanalyzing exome cases unsolved in the first round diagnosis.14 The second data set came from a study demonstrating the better performance of clinical expert–driven pipeline than other computational methods.15 In the clinical expert–driven pipeline, clinical experts first manually curated a candidate gene list based on the phenotypes of a patient, then, pathogenic variants were selected from within the gene list through manual evaluation of the results from a set of computational prediction tools.
Code availability
The Xrare package is available as an easy-to-install docker image at https://web.stanford.edu/~xm24/Xrare/.
RESULTS
ERIC score is robust to phenotype imprecision and noise
A robust phenotype similarity score should have two main features: (1) imprecise/ancestral phenotypes should lead to small changes in scores; (2) scores between random noise phenotypes should be small. Resnik similarity is defined as the IC of MICA, and ERIC reduces to Resnik similarity when two phenotype terms have an ancestor–descendant relationship (e.g., phenotype A and B in case I of Fig. 2a). Therefore, both ERIC and Resnik should be more robust to phenotype imprecision than JC and Lin, which have a penalty to imprecise phenotypes based on the definitions of JC and Lin (see “Materials and methods”). However, Resnik cannot differentiate the similarities with ancestral (case I) from neighboring (case II) phenotypes (Fig. 2a). The JC similarity considers only the IC distance between two phenotypes and no usage of the IC of MICA, thus it fails to differentiate deep phenotypes (case II) from shallow phenotypes with broad MICA (case III in Fig. 2a). Noise phenotypes are frequently unrelated, far from the true phenotypes, such as phenotype E relative to A in case IV of Fig. 2a. The Lin, Resnik, and JC scores are small values between the noise phenotype A and E, which could still have a small residual effect on the final performance of phenotype similarity. However, ERIC has a built-in zero cut-off such that phenotype pairs outside of certain distance are considered unrelated. In summary, these design principles of ERIC make it robust against both noise and imprecision.

Case illustrations and performance evaluation of Emission-Reception Information Content (ERIC) score. a Case illustrations demonstrating ideas of ERIC score. Four different phenotype cases (I to IV) are shown as examples. A–E are phenotype terms in the phenotype ontology tree, and R is the root. A and B are phenotype terms in comparison. C is the most informative common ancestor (MICA) of phenotypes A and B in cases I, II, and III. In case IV, both phenotypes A and E are far from the outside of the circle with the center of D (the MICA of A and E) and the radius of IC(D), thus E is considered as a noisy phenotype of A by ERIC score. b Performance evaluation of phenotype similarity measures for prioritizing target genes. Resnik, JC, Lin, and ERIC scores were compared. We simulated phenotypes with various imprecision and noise levels, e.g., 2|2|4 represents two true precise phenotypes in the Human Phenotype Ontology (HPO) database, two imprecise phenotypes, and four random noise phenotypes. The top panel represents phenotype sets simulated with imprecision. The middle panel represents phenotype sets simulated with noise. The bottom panel represents phenotype sets with both imprecision and noise. The x-axis is the rank percentile of target genes among 8643 OMIM genes. The y-axis is the cumulative distribution function (CDF) of the rank percentile.
To evaluate the efficiency of ERIC score, we simulated HPO phenotypes of patients associated with target genes (see patient phenotype simulations in Supplementary Methods) and calculated the ranks of target genes in the whole genome using ERIC and three existing alternative methods: Resnik,20 Lin,21 and Jiang-Conrath22 (JC). When imprecise phenotypes are included, ERIC and Resnik are more accurate than JC and Lin (Fig. 2b). When phenotypes are mixed with noise, Resnik’s performance deteriorated dramatically to a level lower than those of JC and Lin, while ERIC remains the best performing method. With both imprecise and noisy phenotypes, as expected in real clinical situations, ERIC offers substantial improvement over existing alternatives. For example, ERIC had approximately 10% more genes than Resnik, JC, and Lin ranked at the top 1%.
Ranking candidate diseases based on a set of clinical phenotypes before genotype information is obtained is usually the first step of rare disease diagnosis. Thus, we carried out an evaluation of ERIC’s performance in phenotype-only disease diagnosis using data from DDG2P (Developmental Disorders Genotype–Phenotype Database, https://decipher.sanger.ac.uk). The data set consists of 1300 diseases and 24,743 HPO pheno–disease associations from clinical samples. When simulating the phenotypes, we extracted 50% of true HPO phenotypes in the data set, added various levels of noise and imprecision, and then compared the rankings of true diseases among 7936 OMIM diseases. Similar to the pheno–geno simulation results in Fig. 2b, we observed substantial performance reduction in the Resnik, JC, and Lin methods when phenotype noise is present (Supplementary Figure S3), while ERIC remains relatively robust. For example, when 1.5× noise is present, ERIC has more than 13.8%, 23.3%, and 25.7% more diseases ranked at top 5 than JC, Lin and Resnik respectively. When 1× noise and 50% imprecision are present in the phenotype set, ERIC still has 49.3% diseases ranked at top 5, while JC, Lin, and Resnik only have 35.7%, 29.4%, and 30.9% ranked at top 5, respectively.
Performance evaluation on known disease genes without inheritance mode
We first evaluated the performance on known disease genes without specifying the inheritance mode. Consistent with previous studies,10,11,12 phenotype-driven methods such as Xrare and Exomiser are far more effective than genotype-only methods CADD, REVEL, M-CAP, and MutationTaster in prioritizing known disease genes (Fig. 3a; more comprehensive comparison of genotype-only methods in Supplementary Figure S4). Among existing phenotype-driven methods (brief method comparison in Supplementary Table 5), Exomiser hiPHIVE model has the best performance. On the other hand, Xrare performs even better and ranks 23% more causal variants at the top (i.e., top 1 rank) than the Exomiser hiPHIVE algorithm. Wilcoxon signed-rank test for rank numbers also demonstrated that Xrare has significantly smaller rank numbers for known genes than Exomiser hiPHIVE (P < 1.9e-15). We noticed that ACMG score (Xrare_ACMG), simply using the ACMG/AMP guideline evidence and combining them into a single weighted score, performs better than other computational genotype-only scores. This suggests the good power of clinical diagnosis guideline-based features in evaluating pathogenic variants of known rare disease genes. We also noticed that a method using only phenotypic feature based on ERIC score, Xrare_ERIC_RP, is better than Exomiser, suggesting the advantage of our ERIC phenotype scoring method.

Performance evaluation on synthetic genomes of patients from various simulation scenarios. a, b Without specifying inheritance mode; (c, d) specifying inheritance mode; (a, c) for known genes; (b, d) for novel genes. Phen-Gen was not included in (a) and (b) for comparison because it required specifying inheritance mode. Lines with triangle dots represent methods utilizing both phenotype and genotype information (Xrare, Xrare_ERIC_RP, Exomiser PhenIX, Exomiser_hiPHIVE, eXtasy, and Phen-Gen), while lines with round dots represent prediction methods using only genotype information (Xrare_ACMG, MutationTaster, CADD, and REVEL). Xrare_ERIC_RP and Xrare_ACMG represent the Xrare model using only the phenotype feature ERIC_RP and only the genotype feature ACMG_score, respectively. CDF cumulative distribution function.
By investigating the performance in different phenotype simulation conditions, we found that Xrare showed better performance than Exomiser in almost every condition, especially when the phenotypic information is imprecise and the interference of phenotypic noise is high. For example, Xrare shows 14% and 21% advantage over Exomiser hiPHIVE and PhenIX at top 1 ranking for the phenotype condition with one precise, two imprecise and two noisy HPO terms, respectively (Supplementary Figures S5). To look further into the improvement by ERIC, we compared the most important feature ERIC_RP with the phenotype scores calculated by Exomiser. ERIC_RP alone shows consistently better performance (Supplementary Figure S6), confirming again the usefulness of ERIC-based score in clinical settings.
Performance evaluation on novel genes without inheritance mode
Next, we evaluated the diagnostic performance of our method when the causative gene is a novel gene, namely a gene that has not been associated with specific phenotypes in the databases. It is seen that again Xrare has the best performance among all methods. For example, Xrare has significantly higher ranks than Exomiser hiPHIVE (P < 2.2e-16) and detects 29% more novel genes at the top 10 than hiPHIVE (Fig. 3b). Surprisingly, genotype-only methods generally perform better than existing phenotype-driven methods, suggesting that the strong genotype and phenotype features associated with novel genes are not well utilized by these phenotype-driven methods. It is also observed that Xrare with ACMG score alone already performs quite well, reinforcing the point that the guideline-based features are extremely useful in evaluating the pathogenicity status of variants for novel genes.
Performance evaluation with known inheritance mode
We also evaluated the performance when the inheritance mode is explicitly known for a patient. Here we included another method, Phen-Gen, in comparison, which requires inheritance mode as input. Xrare showed consistently higher efficiency than Exomiser and Phen-Gen in the dominant and recessive model on known (Fig. 3c) and novel (Fig. 3d) genes. The poor performance of Exomiser_PhenIX for novel genes is probably because it is solely based on known human phenotype annotations, and thus not suitable for this purpose. There are dramatic performance differences between the recessive and dominant mode, which is likely due to the two-allele requirements for recessive genes. At least two candidate pathogenic alleles (prefiltered by some criteria such as low population allele frequency) in a gene are required in the recessive mode. This would filter out most of the candidate genes (~70–90%) where only single candidate allele can be found in the gene.
Benchmarking on real clinical data sets
Finally, we evaluated the method performance using two recently published real clinical exome sequencing data sets, where the HPO-encoded phenotypes of individual patients and causal pathogenic variants are provided. The first data set consists of 45 clinically confirmed variants from a study reanalyzing exome cases unsolved in the first round diagnosis.14 Thus testing on this data set will allow us to assess the performance of the methods in hard-to-solve clinical exome cases. We found that Xrare has a 32% advantage over hiPHIVE in detecting known genes at the top 5 ranking and a 40% advantage for novel genes (Fig. 4a). The next data set consists of 59 confirmed causal variants from a recent study reporting the performance of a clinically driven pipeline.15 Consistent with the reported findings, the clinical expert–driven pipeline outperformed existing methods such as Exomiser_PhenIX, Exomiser_hiPHIVE, MutationTaster, and CADD. It also performs better than Xrare model based only on phenotype ERIC-score or only on ACMG score. However, the full Xrare model performs significantly better than even the clinical expert–driven pipeline, for example, it predicts almost 15% more causative variants than the clinical expert–driven pipeline as the top 1 gene (Fig. 4b).

Performance evaluation on two real exome sequencing clinical data sets. a Hard-to-solve clinical cases14 with known genes on the left panel and novel genes on the right panel. b Data used in performance evaluation of clinical expert–driven pipeline.15 Lines with triangle dots represent methods utilizing both phenotype and genotype information, while lines with round dots represent prediction methods using only genotype information. CDF cumulative distribution function.
DISCUSSION
We presented a new machine learning method for variant prioritization. First, a new phenotype similarity measure was developed to handle the imprecise and noisy nature of clinical phenotypes, and a gradient tree boosting approach was used to extend gene–phenotype annotations to genes not yet annotated in HPO. Second, to leverage current best practice in clinical genetics, we defined and included ACMG/AMP guideline–based genetic features for the training of our model. However, instead of using a fixed decision tree to classify variants as in the current guideline, we used a gradient tree boosting approach to combine all genotypic and phenotypic features to predict the pathogenicity of a variant. Based on the comprehensive evaluation of simulation experiments and real clinical data, our method shows significantly better performance than all previous methods, including the clinical expert–driven methods, regardless of whether the genes contributing to the genetic disease are known or novel.
Our results demonstrated the importance of phenotype score and clinical guideline features. ERIC_RP and ACMG_score are the first and second most important features for prediction in the model, respectively (Fig. 1b). For known genes, phenotype-driven methods such as Exomiser, Phen-Gen, and Xrare all have good performances. Especially in real clinical applications, phenotype-driven methods were generally demonstrated to be more efficient and powerful than other methods.15,24 The 2015 ACMG/AMP guideline was an important milestone for the clinical genetics community because it provided a framework for major evidence categories needed for Mendelian disease diagnosis. Thus even a simple ACMG score integrating the guideline evidences (Xrare_ACMG) had a decent performance for both known and novel genes. Incorporating more refined score schema (e.g., Sherloc25) could potentially further enhance the performance of our model.
One common caveat in supervised machine learning methods is the overestimate of performance due to unexpected additional information in the training data leaking into the evaluation data. We used several approaches to prevent data leakage in model training and performance evaluation. First, we isolated sets of background or negative variants for different purposes: feature calculation (1000 Genomes, ESP, and ExAC), training (UK10K), and evaluation (400 independent whole genomes). Second, we split pathogenic variants by their publishing years. We found random splitting, as done in many cross-validation style model training, significantly reduces the performance in prioritizing novel genes (Supplementary Table S6 and S7). Although random splitting shows even better performance than year splitting for known genes because of the larger number of training data, we observed a dramatic decrease in novel genes (e.g., 57% vs. 4% ranked at top 1). Third, we carefully chose the existing prediction scores learned from known pathogenic variants. For example, we found that the M-CAP scores of ClinVar variants showed very different distributions across years (Supplementary Figure S2). Thus M-CAP is excluded from our features. Fourth, during evaluation, human phenotypes annotated to a gene firstly identified in 2013–2015 were removed from our phenotype annotation database to ensure that phenotypes of the gene are truly unknown at the time. Fifth, since the gene–phenotype associations extracted from rare human diseases were downloaded from HPO in 2016, we removed all phenotypes of novel genes in the data set to ensure that the phenotypes of novel genes in rare human diseases remain unknown.
There are a few limitations to our method. First, our current model is focused on the HPO and UPO phenotype annotations. The Monarch Initiative26 and HPO team are continuing the development of HPO and integration of other model organism phenotypes, which will keep improving the diagnostic rate of HPO based methods. Thus the model should be extended when more comprehensive phenotype annotations are available. For example, incorporating more expert-curated phenotype databases such as inborn error metabolism database IEMbase27 into our model could potentially further increase the diagnostic yield for specific disease areas. Second, our current model is useful mainly for the prioritization of variants near coding regions. Genome sequencing usually led to increased rare disease diagnostic rate in clinics,28,29 yet the identification of pathogenic noncoding variants is a big challenge. Because the vast majority of ClinVar pathogenic variants are in gene coding regions, our model would be less powerful for noncoding regulatory variants, even though phenotype score and a few regulatory function prediction features like CADD were included. Methods like Genomiser30 could be an inspiration in this area. An extension of our model with more noncoding pathogenicity prediction scores (e.g., EIGEN,31 LINSIGHT32) and integrated regulatory information33 of gene expression, chromatin accessibility from ENCODE and Roadmap Epigenomics data34 could allow better prioritization of all coding and regulatory variants in Mendelian diseases.
References
Boycott KM, Rath A, Chong JX, et al. International cooperation to enable the diagnosis of all rare genetic diseases. Am J Hum Genet. 2017;100:695–705.
Posey JE, Harel T, Liu P, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. 2017;376:21–31.
Taylor JC, Martin HC, Lise S, et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet. 2015;47:717–726.
Chong JX, Buckingham KJ, Jhangiani SN, et al. The genetic basis of Mendelian phenotypes: discoveries, challenges, and opportunities. Am J Hum Genet. 2015;97:199–215.
Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315.
Jagadeesh KA, Wenger AM, Berger MJ, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48:1581–1586.
Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99:877–885.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291.
Sifrim A, Popovic D, Tranchevent L-C, et al. eXtasy: variant prioritization by genomic data fusion. Nat Methods. 2013;10:1083–1084.
Robinson PN, Köhler S, Oellrich A, et al. Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res. 2014;24:340–348.
Javed A, Agrawal S, Ng PC. Phen-Gen: combining phenotype and genotype to analyze rare disorders. Nat Methods. 2014;11:935–937.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Eldomery MK, Coban-Akdemir Z, Harel T, et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017;9:26.
Stark Z, Dashnow H, Lunke S, et al. A clinically driven variant prioritization framework outperforms purely computational approaches for the diagnostic analysis of singleton WES data. Eur J Hum Genet. 2017;25:1268–1272.
Friedman JH Greedy function approximation: a gradient boosting machine. Ann Stat. 2001;29:1189–1232.
Chen T, Guestrin C. XGBoost: a scalable tree boosting system. In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. KDD ’16. New York, NY: ACM; 2016:785–794.
Köhler S, Vasilevsky NA, Engelstad M, et al. The Human Phenotype Ontology in 2017. Nucleic Acids Res. 2017;45(D1):D865–D876.
Köhler S, Doelken SC, Ruef BJ, et al. Construction and accessibility of a cross-species phenotype ontology along with gene annotations for biomedical research. F1000Res. 2013;2:30.
Resnik P. Using information content to evaluate semantic similarity in a taxonomy. In: Proceedings of the 14th International Joint Conference on Artificial Intelligence. Vol. 1. IJCAI ’95. San Francisco, CA: Morgan Kaufmann Publishers; 1995:448–453.
Lin D. An information-theoretic definition of similarity. In: Proceedings of the Fifteenth International Conference on Machine Learning. ICML ’98. San Francisco, CA: Morgan Kaufmann Publishers; 1998:296–304.
Jiang JJ, Conrath DW. Semantic similarity based on corpus statistics and lexical taxonomy. In: Proceedings of the 10th Research on Computational Linguistics International Conference. Taipei, Taiwan: The Association for Computational Linguistics and Chinese Language Processing (ACLCLP); 1997:19–33.
Wang JZ, Du Z, Payattakool R, Yu PS, Chen C-F. A new method to measure the semantic similarity of GO terms. Bioinformatics. 2007;23:1274–1281.
Bone WP, Washington NL, Buske OJ, et al. Computational evaluation of exome sequence data using human and model organism phenotypes improves diagnostic efficiency. Genet Med. 2016;18:608–617.
Nykamp K, Anderson M, Powers M, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017;19:1105–1117.
Mungall CJ, McMurry JA, Köhler S, et al. The Monarch Initiative: an integrative data and analytic platform connecting phenotypes to genotypes across species. Nucleic Acids Res. 2017;45(D1):D712–D722.
Lee JJY, Wasserman WW, Hoffmann GF, van Karnebeek CDM, Blau N. Knowledge base and mini-expert platform for the diagnosis of inborn errors of metabolism. Genet Med. 2018;20:151–158.
Stavropoulos DJ, Merico D, Jobling R, et al. Whole genome sequencing expands diagnostic utility and improves clinical management in pediatric medicine. NPJ Genomic Med. 2016;1:15012.
Bick D, Fraser PC, Gutzeit MF, et al. Successful application of whole genome sequencing in a medical genetics clinic. J Pediatr Genet. 2017;6:61–76.
Smedley D, Schubach M, Jacobsen JOB, et al. A whole-genome analysis framework for effective identification of pathogenic regulatory variants in Mendelian disease. Am J Hum Genet. 2016;99:595–606.
Ionita-Laza I, McCallum K, Xu B, Buxbaum JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet. 2016;48:214–220.
Huang Y-F, Gulko B, Siepel A. Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat Genet. 2017;49:618–624.
Duren Z, Chen X, Jiang R, Wang Y, Wong WH. Modeling gene regulation from paired expression and chromatin accessibility data. Proc Natl Acad Sci USA 2017;114:E4914–E4923.
Kundaje A, Meuleman W, et al. Roadmap Epigenomics Consortium, Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330.
Acknowledgements
We would like to thank S. Köhler for feedback on HPO. This work was supported by National Institutes of Health (NIH) grants R01HG007834 and P50HG007735.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosure
K.Z., Q.L. and X.M. are employees of GenomCan Inc. W.H.W. is scientific advisor of GenomCan Inc. C.D.B. declares no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Q., Zhao, K., Bustamante, C.D. et al. Xrare: a machine learning method jointly modeling phenotypes and genetic evidence for rare disease diagnosis. Genet Med 21, 2126–2134 (2019). https://doi.org/10.1038/s41436-019-0439-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0439-8
Keywords
This article is cited by
-
Is artificial intelligence for medical professionals serving the patients?
Systematic Reviews (2024)
-
VUS next in rare diseases? Deciphering genetic determinants of biomolecular condensation
Orphanet Journal of Rare Diseases (2024)
-
Medical-informed machine learning: integrating prior knowledge into medical decision systems
BMC Medical Informatics and Decision Making (2024)
-
Refined preferences of prioritizers improve intelligent diagnosis for Mendelian diseases
Scientific Reports (2024)
-
Phenotypic similarity-based approach for variant prioritization for unsolved rare disease: a preliminary methodological report
European Journal of Human Genetics (2024)
