
Abstract
Purpose
Several genes on hereditary breast and ovarian cancer susceptibility test panels have not been systematically examined for strength of association with disease. We employed the Clinical Genome Resource (ClinGen) clinical validity framework to assess the strength of evidence between selected genes and breast or ovarian cancer.
Methods
Thirty-one genes offered on cancer panel testing were selected for evaluation. The strength of gene–disease relationship was systematically evaluated and a clinical validity classification of either Definitive, Strong, Moderate, Limited, Refuted, Disputed, or No Reported Evidence was assigned.
Results
Definitive clinical validity classifications were made for 10/31 and 10/32 gene–disease pairs for breast and ovarian cancer respectively. Two genes had a Moderate classification whereas, 6/31 and 6/32 genes had Limited classifications for breast and ovarian cancer respectively. Contradictory evidence resulted in Disputed or Refuted assertions for 9/31 genes for breast and 4/32 genes for ovarian cancer. No Reported Evidence of disease association was asserted for 5/31 genes for breast and 11/32 for ovarian cancer.
Conclusion
Evaluation of gene–disease association using the ClinGen clinical validity framework revealed a wide range of classifications. This information should aid laboratories in tailoring appropriate gene panels and assist health-care providers in interpreting results from panel testing.
Similar content being viewed by others


INTRODUCTION
The use of gene panel testing for determining hereditary cancer risk has dramatically increased within the last few years. This increase in panel testing uptake is largely driven by the convenience and cost-effectiveness of having tests that include all genes associated with each type of hereditary cancer.1,2,3 There is also the concern of missing a possible hereditary form of cancer in a patient who may have a less typical presentation for a given cancer syndrome and/or missing family history data to guide testing decisions.3,4,5,6 In addition, there is increasing use of germline status to make decisions about treatment of advanced cancers.
However, casting a wide diagnostic net has nontrivial consequences. Larger gene panels increase the likelihood that a patient may receive an inconclusive result. This could include variants of uncertain significance (VUS) or a presumed pathogenic variant in a lower penetrance gene that does not explain, or fully explain, the family history.7,8,9 The lack of established cancer risk estimates for some of these commonly tested genes is also a drawback, although some groups are working to improve this knowledge.7,8 This also means that medical management guidelines are scant, or nonexistent, for some of these lower, moderate risk and/or less well-characterized genes.8,9
The National Comprehensive Cancer Network (NCCN) recommends that multigene test panels be considered when a patient’s personal and/or family history is suggestive of a hereditary cancer syndrome.10 However, NCCN guidelines note that many of the genes on hereditary breast and ovarian cancer susceptibility panels have not been systematically examined for their strength of association with disease and do not have specific medical management recommendations.10 Additionally, many genes are grouped together on pan-cancer or breast and ovarian cancer panels although a number of those genes have only been associated with either breast cancer or ovarian cancer. When a pathogenic variant is identified, it can be difficult for health-care providers to know the strength of evidence of disease risk and whether the variant confers an elevated risk of breast cancer, ovarian cancer, or both for their patient.
The Clinical Genome Resource (ClinGen) is a National Institutes of Health (NIH)-funded program dedicated to creating publicly available data that assesses the clinical relevance of genes and variants within specific diseases.11 ClinGen developed a clinical validity framework that facilitates systematic evaluation of publicly available evidence to assign the strength of the evidence for the association of a gene with a disease into one of the following clinical validity classifications: Definitive, Strong, Moderate, Limited, No Reported Evidence or Conflicting Evidence Reported, Disputed, or Refuted.12 We describe the application of this framework to evaluate 31 genes currently on clinical testing panels in predisposition to breast cancer, ovarian cancer, or complex syndromes involving these cancers.
MATERIALS AND METHODS
The ClinGen Breast/Ovarian Gene Curation Expert Panel (GCEP), including a molecular geneticist, three postdoctoral fellows, two research scientists, two laboratory-based genetic counselors, and two clinical-based genetic counselors specializing in hereditary cancer, was convened to address the varying levels of evidence available in the medical literature linking particular genes with inherited predisposition to develop breast and/or ovarian cancer. The GCEP initially generated a list of genes that are commonly found on clinically available breast and/or ovarian cancer or pan-cancer panels from multiple testing laboratories or that are associated with hereditary breast and/or ovarian cancer in OMIM in July 2015 (ref. 13). The GCEP along with other experts on the ClinGen Hereditary Cancer Clinical Domain Executive Committee, comprised of medical geneticists, medical oncologists, molecular geneticists, scientists, and genetic counselors, then prioritized genes for curation that were common to most panels, as well as genes on panels that had a more controversial association with breast and/or ovarian cancer.
The GCEP finalized a list of 31 high-priority genes for curation using the ClinGen Gene Curation framework.12 In the case of MUTYH, we curated this gene for the autosomal dominant and recessive risk separately, which led to 32 possible gene/disease associations. Of note, we curated monoallelic but not biallelic pathogenic variants in mismatch repair (MMR) genes due to the lack of reports suggesting the association with breast and/or ovarian cancer. If the gene was associated with a genetic syndrome for which breast or ovarian cancer was a known feature in OMIM, and likely part of diagnostic criteria, the gene was curated for the syndrome and not isolated, or familial, cancer risk. The ClinGen Clinical Validity Framework was not designed to parse out risk of individual features of a genetic syndrome. If a gene was not associated with a particular syndrome, the gene was curated for its familial risk of breast and ovarian cancer. For instance, STK11 was curated for Peutz–Jehgers syndrome, for which breast cancer is a known feature, and the TP53 gene was curated for familial ovarian cancer instead of Li–Fraumeni syndrome.
The Gene Curation framework takes into account genetic evidence, through publications describing patients evaluated for the gene in question, including case reports, family studies, and case–control association studies. The framework also includes experimental evidence linking genes to disease through biochemical function; protein interaction; protein expression; functional alterations in cells, animals, or patients; and knockout/-in and/or phenotypic rescue data. A maximum of 12 points are assigned for genetic evidence and 6 points are assigned for experimental evidence for a combined maximum total of 18 points. Of note, the clinically validity classifications do not correlate with the risk of cancer when harboring a pathogenic variant. For example, a Moderate gene association does not imply a moderate risk of cancer. The No Reported Evidence classification was applied to gene/disease associations where there was no case–control or case-level data that qualified for scoring. A Disputed classification simply indicates that there are conflicting data and/or opinions regarding the association. The Refuted classification was assigned only when multiple well-powered case–control studies clearly indicated an odd ratio (OR) close to or below 1.0 and the contradicting evidence outweighed any supporting evidence. In some cases, there were actual claims that a particular cancer was not associated with a specific gene, and that information was used as disputing or refuting evidence.
Gene biocurators were trained on version 4 of the ClinGen Gene Curation Standard Operating Procedure (SOP).14 Two biocurators were assigned a particular gene and asked to query the literature for associations with breast cancer and separately with ovarian cancer. A two-biocurator system was used to help ensure that all relevant publications were reviewed. Biocurators used PubMed and OMIM resources for literature searches. Curation results were discussed during bimonthly GCEP meetings and provisional classifications were assigned. Classifications were finalized only after assertions were approved by the ClinGen Hereditary Cancer Executive Committee.
Three main types of genetic evidence were used: case–control studies, segregation data, and case reports, and several adjustments were made to the ClinGen scoring system as the cancer curation process proceeded. Specifically, although the ClinGen clinical validity framework SOP allows for scoring of individual probands with the disease of interest and a suspected pathogenic variant within the gene of interest, we found that case–control studies were more appropriate to use than case-level data given the prevalence of breast cancer in the general population and the large numbers of individuals being tested for variants within these genes. Therefore, whenever case–control data was available, case reports were not scored because they posed the highest risk of spurious findings. However, case reports in which segregation data were available were scored as recommended by the SOP. To include case–control association studies, we required an OR (or a similar/equivalent metric) of 2.0 or greater with a significant P value (<0.05) and confidence intervals not crossing 1 in support of an association with breast cancer. Because ovarian cancer is a relatively rare cancer in the general population with less risk of phenocopies, studies with OR >1.5 were included in scoring for each gene. If neither case–control studies nor segregation data were available, each proband from a case report was given a score of 0.1, which was downgraded from the default score of 0.5–1.5. Importantly, we required these probands to have a suspected loss-of-function (LoF) variant with substantial functional data demonstrating a deleterious effect in order to be counted. We excluded any variants that were consistent with a classification of benign, likely benign, or VUS in ClinVar, and variants that had limited evidence of pathogenicity or high minor allele frequencies in ExAC or gnomAD.15,16 No adjustments to the recommended scoring matrix were made for experimental evidence. Although some overlapping evidence exists, experimental evidence was curated separately for breast and for ovarian cancer to ensure that the information was specific and robust for each cancer.
After curations were complete, six breast/ovarian or pan-cancer panels listed in the Genetic Test Registry were sampled to determine how the genes on those panels were evaluated in the curation process (e.g., Definitive, Moderate, etc.).17
RESULTS
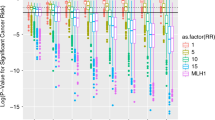
Review of existing hereditary breast and ovarian cancer panels and OMIM identified 31 genes to be curated using the ClinGen clinical validity matrix, and a final assertion is provided based on genetic and experimental evidence reviewed from literature searches (Table 1). As mentioned previously, there are a total of 32 gene–disease curation pairs for ovarian cancer due to the MUTYH gene being curated separately for autosomal dominant and recessive risk. However, there are only a total of 31 gene–disease curations for breast cancer because we were unable to score NF1 for breast cancer susceptibility. While the NF1 gene may confer an increased risk of breast cancer in young women with a clinical diagnosis of neurofibromatosis type 1 based on case–control studies, most of the women in the literature were not genotyped. Instead, the diagnosis was based on clinical criteria, and therefore, we decided not to score this gene with the clinical validity framework, which assumes a molecular evaluation has been performed.18,19,20,21,22,23 The resulting gene–disease assertions are displayed in Fig. 1. Full details for all gene curation results can be found and downloaded from the ClinGen website.24

(a) Clinical validity classifications of 31 gene–disease pairs associated with syndromic or familial breast cancer susceptibility. Consensus genetic and experimental evidence scores are depicted for each gene–disease relationship wherein a Limited preliminary classification scored 0.1–6 total points, a Moderate preliminary classification had 7–11 total points, a Strong preliminary classification scored 12–18 total points, and a Definitive classification scored 12–18 total points and achieved replication over time (indicated by r/t). (b) Clinical validity classifications of 32 gene–disease pairs associated with syndromic or familial ovarian cancer susceptibility.
Definitive assertions were made for 10/31 (32.3%) gene–disease pairs for breast and 10/32 (31.3%) for ovarian cancer. Moderate assertions were made for only 2 genes: RECQL for breast cancer and PALB2 for ovarian cancer. Several genes fell into the Limited range: 6/31 (19.4%) for breast and 6/32 (18.8%) for ovarian cancer. There were only 2 genes (SLX4 and XRCC2) for which only case-level data were available that required a downgrade to 0.1 points per proband, and both genes scored in the Limited range for breast cancer. Despite selecting genes that appear on clinical hereditary panels, we determined Disputed or Refuted assertions for 9/31 (29%) genes for breast and 4/32 (12.5%) for ovarian cancer. No Reported Evidence of disease association was asserted for 5/31 (16.1%) genes for breast and 11/32 (34.4%) for ovarian cancer. The higher percentage for ovarian cancer is likely due to the primary breast cancer association for most genes. Similarly, genes with Disputed or Refuted evidence for one cancer were typically classified as Limited or a higher assertion for the other cancer, simply reflecting that their role on cancer panels was likely due to their primary association with the other cancer. However, there were three exceptions. The MRE11A gene was disputed for both breast and ovarian cancer. GEN1 and RINT1 had disputed evidence for breast cancer and No Reported Evidence for ovarian cancer. The breakdown of gene assertions for breast cancer and ovarian cancer is displayed in Fig. 2a, b, respectively.

(a) Thirty-one gene–disease pair curations were assessed for their strength of evidence with syndromic and familial breast cancer susceptibility. MUTYH was assessed for autosomal dominant and recessive risk associations, which counted as two separate curations. NF1 was unable to be scored due to lack of genotyping in affected individuals in the literature, which resulted in 31 gene–disease curation pairs. (b) Thirty-two gene–disease pair associations were assessed for ovarian cancer susceptibility. MUTYH was assessed for autosomal dominant and recessive risk associations and NF1 was assessed for familial ovarian cancer susceptibility resulting in 32 gene–disease curation pairs. (c) Displays the number of genes on panels by their highest clinical validity classification to highlight the number of genes with less than Definitive classifications on testing panels. Each gene is represented by its highest clinical validity score, be it for breast or ovarian cancer. NF1 was seen on 3 of 6 test panels but not included above. This gene is likely on panels due to its reported association with breast cancer. Because we were unable to score its association with breast cancer, the gene was not included to avoid using the No Reported Evidence assertion for familial ovarian cancer, which seems to misrepresent its importance on gene panels. (d) Gene panels were assessed in February 2018. Again, NF1 is not represented for the same reason as specified above.
These results were used to evaluate the classification breakdown of genes on six available testing panels as shown in Fig. 2c, d. To account for the different levels of evidence between the two cancers, we used the highest classification for each gene to highlight the number of Definitive, Moderate, Limited, Disputed, and No Reported Evidence genes that are available for testing on these panels. All genes in the Definitive range for one or both cancer types were offered by all six laboratories, with the exception of STK11, which a number of laboratories removed from their panel because of the absence of pathogenic STK11 variants after testing a large number of individuals referred for hereditary breast and ovarian cancer testing. This reflects that a gene may have an increased risk of cancer within a well-described syndrome but account for a very small proportion of the overall number of individuals at genetic risk for these cancers. Most of the genes in the Disputed or No Reported Evidence range were only available on one or two panels. Genes scoring in the Limited range were generally on few panels, with the exception of NBN, which is offered on all six panels.
DISCUSSION
We used the ClinGen clinical validity framework to assess the gene–disease relationships between genes commonly found on hereditary breast and ovarian cancer panels. The majority of genes fell into the Definitive range (18/31; 58.1%) for one or both types of cancer. All of the genes associated with syndromic cancer risk fell into the Definitive range. BRCA1 and BRCA2 were the only genes with a Definitive assertion for predisposition to both breast and ovarian cancers. Of the familial risk genes analyzed, five fell into the Definitive range for breast cancer (ATM, BARD1, CDH1, CHEK2, and PALB2) and three fell into the Definitive range for ovarian cancer (BRIP1, RAD51C, and RAD51D). There were a handful of genes that did not demonstrate an association with either cancer or had a Disputed association with both cancers but were on at least one hereditary cancer panel (CHEK1, MRE11A, PIK3CA, and RINT1).
Most genes on testing panels from the six laboratories surveyed fall into the Definitive range for breast and/or ovarian cancer. Only two laboratories had genes that were Disputed and none had genes that were Refuted for both types of cancer. The genes scoring in the Limited range were observed on an average of 2 panels. In contrast, NBN had Limited support as a breast or ovarian cancer gene, but was found on all six commercial gene panels. Early reports suggested an association with breast cancer risk, but more recent case–control studies have not demonstrated that association.25,26,27,28 The UK Cancer Genetic Group recently published that they found little evidence of the association with breast or ovarian cancer and recommended that the NBN gene not be included on hereditary cancer gene panels in the United Kingdom.29 Our curation results suggest, and experts agreed, that this gene should remain in the Limited range for now, although it was noted that as larger case–control studies become available this gene–disease pair could eventually be Disputed. The CHEK1 gene, for which we found no reported evidence for breast or ovarian cancer risk, was offered on one pan-cancer test panel. The MCPH1 gene was not found on a panel listed on the Genetic Test Registry.17
One surprising curation result was the Moderate determination for RECQL and breast cancer. This was one of the first genes in our curation process that alerted us to the substantial difficulties posed by phenocopies and assigning points to individual probands. Cybulski et al. and Kwong et al. published several probands with presumed LoF variants in individuals with breast cancer, which would score RECQL in the Definitive range if the default matrix scoring was used.30,31 Even with the adjustments we made to scoring for case-only studies, this gene scored well. This result may either confirm RECQL as a cancer risk gene, or highlight a limitation of the matrix for cancer predisposition genes. The fact that this gene is offered on only 1 of 6 commercial panels surveyed suggests a lack of confidence by many commercial laboratories regarding a role for RECQL in breast cancer. Alternatively, absence from panels means data for large case–control data sets are not being accumulated from large commercial laboratories. Despite this concern, we were unable to find any publications that provided negative gene association data or questioned RECQL as a cancer predisposition gene.
None of the genes evaluated for breast or ovarian cancer were classified in the Strong range. The Strong range is assigned to genes that score 12–18 points where the gene–disease association has not been established over a period of greater than 3 years. By design, genes will fall into this classification only temporarily because by the time there is enough evidence to score into the Strong range, enough time may also have passed to classify the gene in the Definitive range.
Several genes fell into the Limited classification range. Genes that have been newly described and are not as well studied may not have the amount of genetic evidence points to score into a higher range. We expect that genes in this category could move to higher classifications over time. In particular, genes with mostly experimental evidence may benefit from larger case–control studies. Genes associated with a lower penetrance or those in which pathogenic variants are extraordinarily rare are also at a disadvantage in this scoring system. Finally, genes that have been erroneously associated with cancer risk may also fall into this classification because there would be little to no ongoing genetic evidence to support their association.
Several curated genes are associated with syndromic forms of cancer risk. Whenever possible we used genetic evidence data for which the proband had the cancer of interest. For instance, STK11 and its association with breast cancer was evaluated using probands with breast cancer who met criteria for Peutz–Jeghers syndrome. We found no cases with suspected STK11 pathogenic variants with malignant epithelial ovarian cancers; however, other types of ovarian tumors, such as sex cord tumors with annular tubules (SCTATs), and other benign tumors are not rare in this disorder.32,33,34,35 Because epithelial ovarian cancer is also not a part of the clinical criteria for Li–Fraumeni syndrome or PTEN hamartoma tumor syndrome, we also examined the strength of evidence associated with the TP53 and PTEN genes and ovarian cancer susceptibility separately; we found Limited and Disputed associations respectively.
In regard to CDH1, all cases with suspected pathogenic variants had lobular breast cancer. Therefore, given the specificity of this subtype of breast cancer and its lower frequency within the general population, we allowed case level data and awarded 1 point/family for LoF variants and used segregation data, which led us to a Definitive classification for lobular breast cancer. We did not find an association between CDH1 suspected pathogenic variants and other subtypes of breast cancer, such as ductal carcinoma, or with ovarian cancer. Even excluding cases with a known personal or family history of gastric cancer, we were able to show a Definitive association with lobular breast cancer risk; hence, this curation is classified as familial lobular breast cancer risk. However, we acknowledge it is possible that all pathogenic variants also infer an increased risk of diffuse gastric cancer.
There has been much controversy over the risk of breast cancer for patients with Lynch syndrome.36,37 This is reflected in Disputed assertions for all of the MMR genes and breast cancer. In contrast to our findings, Roberts et al. recently published a case–control study that demonstrated an association between pathogenic variants in MSH6 and PMS2 and breast cancer.38 While this study did meet our case–control guidelines for scoring, there were other large and recent case–control studies that do not find this association.27,28,39,40 Therefore, the most appropriate designation at this time was Disputed. A Disputed designation does not suggest no association between these genes and breast cancer; it only reflects that the data remain conflicted at this time. We found No Reported Evidence for breast or ovarian cancer risk and the EPCAM gene. This may be due to the limited number of patients with this molecular type of Lynch syndrome. The MMR and EPCAM genes were not curated individually for ovarian cancer because ovarian cancer is a well-established cancer within Lynch syndrome. Hence, the Definitive assertion for ovarian cancer in each of the MMR and EPCAM genes. However, we did observe that there was less case–control evidence for an association of PMS2 or MSH6 and ovarian cancer. Again, this could simply reflect a smaller number of patients with Lynch syndrome having pathogenic variants within one of these two genes or a difference in the cancer risk of these different forms of Lynch syndrome. More research would be needed to address this question.
Throughout this curation process it became apparent that frequent updates, or recurations, would be necessary to account for new data. As new case–control studies were published from large commercial laboratory data sets, we revisited previously curated genes to assess the scoring or potentially move genes into the Disputed/Refuted classifications. Based on this experience, the GCEP has decided to update all curations as they appear on www.clinicalgenome.org on at least an annual basis, or as needed based on newly published case–control data sets.
A potential limitation of risk assessment models based on familial ascertainments results from misclassification of phenocopies with the risk allele under analysis. We attempted to lessen the effect by not using case-level data when stronger lines of evidence, like case–control studies, were available. This made it more difficult for a gene to score in the Definitive range simply because breast cancer is relatively common and individuals with cancer frequently undergo large panel testing. We may find that we need to continue to adjust our scoring system to accurately reflect available genetic evidence. Another obvious challenge is that pertinent data could have been inadvertently overlooked. By using two biocurators with expert reviews we hope to have limited this possibility as much as possible, and the ClinGen community is looking into methods to improve data extraction from the literature to lessen this confounding factor. We are also limited by the restricted range of ancestry of patients undergoing hereditary cancer panel testing from which much of the data analyzed is derived.
This work, in contrast to “expert panel” assessments such as NCCN, provides specific context regarding the amount of evidence that is available that links a particular gene to be associated with breast cancer risk, ovarian cancer risk, or a risk for both cancers. Again, this work does not provide an estimation of cancer risk associated with these genes. Future work is needed to carefully look at breast and/or ovarian cancer risk in individuals with pathogenic variants in the genes that seem to have a valid association with cancer. Our work has simply helped to clarify the genes that need a thorough examination of cancer risk. An advantage of this empiric approach is that it is also dynamic; our ClinGen GCEP is committed to updating this online resource on a regular basis. It is important for clinicians to be aware of this resource that indicates that some genes on current panels have limited evidence of association with disease, as this will have important implications for counseling. This could impact a clinician’s decision to act on the presence of a pathogenic variant in a gene with limited association with breast and/or ovarian cancer in their patient. Knowing that larger gene panels are more likely to contain genes of limited or controversial evidence for any given cancer type of interest should affect the clinician’s decision about which panel is most appropriate for their patient. Additionally, being able to prepare patients for the fact that some genes on their test panel have less evidence, and/or less relevance to their particular medical or family history, and therefore, likely limited medical management guidelines may also be important. Because there is often overlap between breast and ovarian cancer gene panels, this work also aids the clinician to determine whether cancer risks are exclusive to one versus the other or for either type of cancer. This work may also encourage commercial laboratories to design cancer gene panels to eliminate genes that have Refuted/Disputed evidence of disease association or limited evidence for all cancer types.
References
Tucker T, Marra M, Friedman JM. Massive parallel sequencing: the next big thing in genetic medicine. Am J Hum Genet. 2009;85:142–154.
Rehm H. Disease-targeted sequencing: a cornerstone in clinic. Nat Rev Genet. 2013;14:295–300.
Stanislaw C, Xue Y, Wilcox WR. Genetic evaluation and testing for hereditary forms of cancer in the era of next-generation sequencing. Cancer Biol Med. 2016;13:55–67.
Rosenthal ET, Bernhisel R, Brown K, et al. Clinical testing with a panel of 25 genes associated with increased cancer risk results in a significant increase in clinical significant findings across a broad range of cancer histories. Cancer Genet. 2017;218-219:58–68.
Kurian AW, Hare EE, Mills MA, et al. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol. 2014;32:2001–2009.
Crawford B, Adams SB, Sittler T, et al. Multi-gene panel testing for hereditary cancer predisposition in unsolved high-risk breast cancer ovarian cancer patients. Breast Cancer Res Treat. 2017;163:383–390.
Easton DF, Pharoah PD, Antoniou AC, et al. Gene-panel sequencing and the prediction of breast-cancer risk. N Engl J Med. 2015;372:2243–2257.
Robson ME, Bradbury AR, Arun B, et al. American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility. J Clin Oncol. 2015;33:3660–3667.
Hall MJ, Forman AD, Pilarski R, et al. Gene panel testing for inherited cancer risk. J Natl Compr Canc Netw. 2014;12:1339–1346.
National Comprehensive Cancer Network (NCCN). Version 1.2018 Genetic/familial high-risk assessment: breast and ovarian. 2017. https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. Accessed 21 February 2018.
Rehm HL, Berg JS, Brooks LD, et al. ClinGen—the Clinical Genome Resource. N Engl J Med. 2015;372:2235–2242.
Strande NT, Riggs ER, Buchanan AH, et al. Evaluating the Clinical Validity of Gene-Disease Associations: An Evidence-Based Framework Developed by the Clinical Genome Resource. Am J Hum Genet. 2017;100:895–906.
OMIM. Hereditary breast and ovarian cancer. https://www.omim.org. Accessed 17 June 2015.
Clinical Genome Resource. Gene clinical validity curation process standard operating procedure version 4. May 2017. https://www.clinicalgenome.org/site/assets/files/8890/gene_curation_sop_2016_version_4_0_7_17_17.pdf. Accessed 21 February 2018.
Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–1067.
Lek M, Karczewski KJ, Minikel EV. Analysis of protein-coding genetics variation in 60,706 humans. Nature . 2016;536:285–291.
Genetic Test Registry. https://www.ncbi.nlm.nih.gov/gtr. Accessed 21 February 2018.
Walker L, Thompson D, Easton D, et al. A prospective study of neurofibromatosis type 1 cancer incidence in the UK. Br J Cancer. 2006;95:233–238.
Wang X, Levin AM, Smolinski SE, et al. Breast cancer and other neoplasms in women with neurofibromatosis type 1: a retrospective review of cases in the Detroit metropolitan area. Am J Med Genet A. 2012;158A:3061–3064.
Seminog OO, Goldacre MJ. Risk of benign tumours of nervous system, and of malignant neoplasms, in people with neurofibromatosis: population-based record-linkage study. Br J Cancer. 2013;108:193–198.
Madanikia SA, Bergner A, Ye X, et al. Increased risk of breast cancer in women with NF1. Am J Med Genet A. 2012;158A:3056–3060.
Sharif S, Moran A, Huson SM, et al. Women with neurofibromatosis 1 are at a moderately increased risk of developing breast cancer and should be considered for early screening. J Med Genet. 2007;44:481–484.
Uusitalo E, Kallionpaa RA, Kurki S, et al. Breast cancer in neurofibromatosis type 1: overrepresentation of unfavourable prognostic factors. Br J Cancer. 2017;116:211–217.
ClinGen. Gene-disease validity. https://www.clinicalgenome.org/curation-activities/gene-disease-validity/results/. Accessed 23 February 2018.
Steffen J, Nowakowska D, Niwinska A, et al. Germline mutations 657del5 of the NBS1 gene contribute significantly to the incidence of breast cancer in Central Poland. Int J Cancer. 2016;119:472–475.
Bogdanova N, Feshchenko S, Schurmann P. et al. Nijmegen breakage syndrome mutation and risk of breast cancer. Int J Cancer. 2008;122:802–806.
Couch FJ, Shimelis H, Hu C, et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. 2017;3:1190–1196.
Kurian AW, Hughes E, Handorf EA, et al. Breast and ovarian cancer penetrance estimates derived from germline multiple-gene sequencing results in women. JCO Precision Oncol. 2017; https://doi.org/10.1200/PO.16.00066. Accessed 27 November 2018.
Taylor A, Brady AF, Frayling IM, et al. Consensus for gene to be including on cancer panel tests in the UK genetics services: guidelines of the UK Genetics Group. J Med Genet. 2018;55:372–377.
Cybulski C, Carrot-Zhang J, Kluzniak W, et al. Germline RECQL mutations are associated with breast cancer susceptibility. Nat Genet. 2015;47:643–646.
Kwong A, Shin VY, Cheuk IWY, et al. Germline RECQL mutations in high risk Chinese breast cancer patients. Breast Cancer Res Treat. 2016;157:211–215.
Howell L, Bader A, Mullassery D, et al. Sertoli Leydig cell ovarian tumour and gastric polyps as presenting features of Peutz-Jeghers syndrome. Pediatr Blood Cancer. 2010;55:206–207.
Li J, Meeks H, Feng BJ, et al. Targeted massively parallel sequencing of a panel of putative breast cancer susceptibility genes in a large cohort of multiple-case breast and ovarian cancer families. J Med Genet. 2016;53:34–42.
Massa G, Roggen N, Renard M, et al. Germline mutation in the STK11 gene in a girl with an ovarian Sertoli cell tumour. Eur J Pediatr. 2007;166:1083–1085.
Resta N, Pierannunzio D, Lenato GM, et al. Cancer risk associated with STK11/LKB1 germline mutations in Peutz-Jeghers syndrome patients: results of an Italian multicenter study. Dig Liver Dis. 2013;45:606–611.
Ford JM. Is breast cancer a part of Lynch syndrome? Breast Cancer Res. 2012;14:110.
Win AK, Lindor NM, Jenkins MA. Risk of breast cancer in Lynch syndrome: a systematic review. Breast Cancer Res. 2013;15:R27.
Roberts ME, Jackson SA, Susswein LR, et al. MSH6 and PSM2 germ-line pathogenic variants implicated in Lynch syndrome are associated with cancer. Genet Med. 2018 Jan 18; https://doi.org/10.1038/gim.2017.254 [Epub ahead of print].
Muller A, Edmonston TB, Corao DA, et al. Exclusion of breast cancer as an integral tumor of hereditary nonpolyposis colorectal cancer. Cancer Res. 2002;62:1014–1019.
Vahteristo P, Ojala S, Tamminen A, et al. No MSH6 mutation in breast cancer families with colorectal and/or endometrial cancer. J Med Genet. 2005;42:e22.
Acknowledgements
We are grateful to members of the ClinGen Hereditary Cancer Executive Committee and the ClinGen Gene Curation Working Group for their expertise and guidance in assessing these gene–disease curation pairs. We also thank Jennifer McGlaughon, Jonathan Berg, and Courtney Thaxton at UNC–Chapel Hill for their thoughtful review and edits of this manuscript. This GCEP is funded by National Human Genome Research & National Cancer Institutes (1U41HG006834, 1U01HG007437, 1U01HG007436, HHSN261200800001E, U41HG009650).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The following authors have made extensive contributions to the literature relative to gene curation and genetic testing: K.O., H.S., and F.J.C. The following authors are an employee, trainee, or consultant for a commercial laboratory that offers hereditary cancer panel testing: N.J.C., K.D., and S.E.P.; N.J.C. and K.D. are employed at GeneDx Laboratories, which provides hereditary cancer panel testing, and S.E.P. consults for the Baylor College of Medicine Molecular Diagnostic Laboratory. The other authors declare no conflicts of interest.
Additional information
Co-first authors Kristy Lee, MS and Bryce A. Seifert.
Rights and permissions
About this article

Cite this article
Lee, K., Seifert, B.A., Shimelis, H. et al. Clinical validity assessment of genes frequently tested on hereditary breast and ovarian cancer susceptibility sequencing panels. Genet Med 21, 1497–1506 (2019). https://doi.org/10.1038/s41436-018-0361-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0361-5
Keywords
This article is cited by
-
EMQN best practice guidelines for genetic testing in hereditary breast and ovarian cancer
European Journal of Human Genetics (2024)
-
Diagnostic yield and clinical relevance of expanded germline genetic testing for nearly 7000 suspected HBOC patients
European Journal of Human Genetics (2023)
-
An overview of genetic services delivery for hereditary breast cancer
Breast Cancer Research and Treatment (2022)
-
High familial burden of cancer correlates with improved outcome from immunotherapy in patients with NSCLC independent of somatic DNA damage response gene status
Journal of Hematology & Oncology (2022)
-
Multigene panel testing for hereditary breast and ovarian cancer in the province of Ontario
Journal of Cancer Research and Clinical Oncology (2021)
