
Abstract
Purpose
TANGO2-related disorders were first described in 2016 and prior to this publication, only 15 individuals with TANGO2-related disorder were described in the literature. Primary features include metabolic crisis with rhabdomyolysis, encephalopathy, intellectual disability, seizures, and cardiac arrhythmias. We assess whether genotype and phenotype of TANGO2-related disorder has expanded since the initial discovery and determine the efficacy of exome sequencing (ES) as a diagnostic tool for detecting variants.
Methods
We present a series of 14 individuals from 11 unrelated families with complex medical and developmental histories, in whom ES or microarray identified compound heterozygous or homozygous variants in TANGO2.
Results
The initial presentation of patients with TANGO2-related disorders can be variable, including primarily neurological presentations. We expand the phenotype and genotype for TANGO2, highlighting the variability of the disorder.
Conclusion
TANGO2-related disorders can have a more diverse clinical presentation than previously anticipated. We illustrate the utility of routine ES data reanalysis whereby discovery of novel disease genes can lead to a diagnosis in previously unsolved cases and the need for additional copy-number variation analysis when ES is performed.
Similar content being viewed by others


Introduction
TANGO2 (transport and Golgi organization 2 homolog) was first identified in 2016 and encodes a protein that is postulated to be involved in redistributing the Golgi membranes into the endoplasmic reticulum (ER). As previously reported, exome sequencing (ES) has identified pathogenic TANGO2 variants in 15 individuals presenting in metabolic crisis with rhabdomyolysis, seizures, progressive intellectual disability, encephalopathy, and cardiac arrhythmias.1,2 As a newly recognized disorder, additional clinical and molecular information is necessary for clinicians and families to recognize and understand the phenotypic spectrum of the disorder. In this collaborative case series, we present clinical and molecular data for 14 previously unreported individuals from 11 unrelated families who harbor compound heterozygous or homozygous TANGO2 variants, expanding the molecular profile and clinical phenotype of TANGO2-related disorder.
Materials and methods
Individuals were initially identified by clinical exome sequencing, gene panel, and/or chromosome microarray that detected pathogenic variants in TANGO2. Families 1–8 underwent ES along with analysis using an in-house copy-number variant (CNV) detection algorithm3 (GeneDx, Gaithersburg, MD). Family 8 had chromosome microarray testing, followed by ES analysis limited to a specific phenotype-driven gene list provided by the ordering clinician that included TANGO2. Family 9 had chromosome microarray testing, followed by reanalysis of exome data (Ambry Genetics, Santa Clara, CA). Family 10 had a chromosome microarray at Kiel (Human Genetics). Family 11 had a chromosome microarray at Kiel (Human Genetics), Germany and ES in Cologne, Germany. This study was approved by the Institutional Review Board at the University of Washington. Informed consent was obtained from all affected individuals or their guardians per the specifications of the respective home institutions, including consent to publish photographs for families 1 and 8. The population frequency of each variant is listed in Table S1.
RNA sequencing
RNA was extracted from patient 1 and control fibroblast cell lines using Trizol (Ambion) and RNA Clean and Concentrator (Zymo Research). RNA sequencing library preparation was performed following the manufacturer's instructions using the TruSeq RNA Library Prep Kit v2 (Illumina), which captures non-strand-specific messenger RNA (mRNA) libraries via poly-A selection. Paired-end 100-bp sequencing was performed on an Illumina HiSeq 4000 with targeted sequencing coverage of 50 M reads. Raw reads were mapped to the human genome (GRCh37/UCSC hg19) using STAR (v.2.2.1). The region surrounding the putative splice site variant NM_152906.6:c.711-3C>G was manually evaluated using the Integrative Genomics Viewer. Sashimi plots were generated using MISO (v.0.5.4).
Patient 1 and control RNA was reverse transcribed following the manufacturer’s instructions using oligo(dT) nucleotides and SuperScript II (Invitrogen). Polymerase chain reaction (PCR) to verify splicing defects was performed using primers designed to amplify regions of TANGO2 encompassing exon 5 through exon 9, and exon 5 through intron 7. GAPDH was used as a loading control. Primer sequences are listed in Table S2.
Results
We describe the clinical and molecular details of 14 individuals with compound heterozygous or homozygous pathogenic variants in TANGO2 from 11 unrelated families (Table 1, Fig. 1a, Table S3). Demographics include 8 females and 6 males with ages ranging from 8 months to 26 years old. Self-declared ancestry included 8/14 of European descent, 2/14 of African American ancestry, 2/14 of Latino ethnicity, 1/14 of European/African American ancestry, and 1/14 of Arab ancestry. In two families, parents were consanguineous.

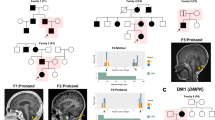
a Pedigrees from 11 families. b Patient photos from family 1 including patient 1 at 2 years 10 months, patient 2 at 2 years, and patient 3 at 9 months. c Patient photos from patient 11 at 13 years.
The most common pathogenic variant in our cohort is the previously described1,2 deletion that encompasses exons 3–9 of TANGO2 (12/22 alleles, 55%). Affected individuals were homozygous for the deletion in five families and compound heterozygous in four families. The remaining pathogenic variants in our cohort have not been previously reported and include a splice site variant (c.711-3C>G), a recurrent nonsense variant (c.94C>T, p.R32*, n = 3), a single-exon 6 deletion (arr[GRCh37] 22q11.21[chr22:20,042,250-20,048,850]), and two missense variants (c.77G>A, p.R26K; c.265G>T, p.G89C) (Fig. 4). We performed RNA expression studies of the c.711-3C>G variant using fibroblasts from patient 1, which showed that intron 8 is retained in the majority of transcripts (Fig. 2). This results in the insertion of 52 amino acids into the protein sequence before ending in a premature stop codon (NM_152906.6 p.R237ins*53). Occasionally, intron 7 is also retained in both control and patient transcripts (data not shown).

NM_152906.6:c.711-3C>G causes aberrant splicing of TANGO2. a Sashimi plot showing TANGO2 transcripts isolated form patient 1 and control fibroblasts. Intron 8 is retained in patient 1 transcripts (*). b Semiquantitative reverse transcription polymerase chain reaction (RT-PCR) validation of aberrant splicing of TANGO2 in patient 1. Primers were designed to amplify exons 5 through exon 9 of TANGO2. The expected product size for a fully spliced transcript is 395 bp, while the expected product size if intron 8 was retained is 1499 bp.
For most individuals, the TANGO2 variants were identified by exome sequencing, often after prior metabolic and genetic testing. Patient 12 was first identified with a heterozygous 22q11 deletion detected by chromosome microarray during a workup for developmental delays. Additional episodic metabolic abnormalities including ketosis, hypoglycemia, and lactic acidosis prompted further analysis of TANGO2, which lies within the 22q11 deletion region. In two additional cases, TANGO2-related disorder was considered in the differential due to previous experience diagnosing an unrelated patient. In two families, the diagnosis was made upon reanalysis of exome data or after the diagnosis was made in a similarly affected younger sibling. In family 1, all three siblings share compound heterozygous variants in TANGO2. The oldest sibling, with a more severe course, also harbors a de novo variant of uncertain significance in UBR5 (c.7554C>A, p.A2585E) that was not present in his siblings. A variant in UBR5 has been previously associated with familial adult myoclonic epilepsy in one family.4
Summary of clinical findings
Initial presentations
Onset of first symptoms ranged from 4 months to 27 months (Table 1, Table S3). The most frequent initial symptom was developmental delay (10/14). Patient 6 presented with a metabolic crisis and concurrent heart failure with dilated cardiomyopathy. Her sister (patient 5), presented at 18 months with speech delay and hypoglycemic generalized tonic–clonic seizures. In the setting of rhinovirus, patient 7 presented with a severe metabolic acidosis, hypoglycemia, hyperammonemia, and elevated creatine kinase at 4 months with acute to subacute left temporal and parietal cortical infarcts on brain magnetic resonance image (MRI). Other presentations include febrile seizures in patient 8 at 18 months with no episode of rhabdomyolysis until age 5 and an episode of ataxia and dizziness at age 2 in patient 10 with subsequent recurrent episodes of encephalopathy, hypoglycemia, and rhabdomyolysis. Patient 11 presented with global developmental delays at 6 months and subsequently developed spasticity and dystonia. His first severe episode of rhabdomyolysis did not occur until 13 years of life.
Phenotypically, individuals are nondysmorphic in appearance with the exception of wide-spaced teeth in patients 1 and 11 and hypodontia in patient 11 (Fig. 1b, c).
Neurologic findings
Seizures are present in 71% (10/14) of individuals, two of which were only related to episodes of hypoglycemia and another to ventricular tachycardia. Seizure types include infantile spasms, myoclonic, tonic, generalized tonic–clonic, partial complex, and multifocal seizures. Three individuals had refractory epilepsy. Brain MRI demonstrated cerebral atrophy in 5/14 patients. Cerebral infarct and/or cytotoxic edema during an acute episode was seen in 3/14 patients. Serial MR images from patient 1 highlight progressive generalized cerebral atrophy that occurred over the course of 1 year (Fig. 3). No structural brain abnormalities were seen in our cohort of patients.

Magnetic resonance imaging (MRI) of patient 1 demonstrating progressive cerebral atrophy. a Axial T2 fluid-attenuated inversion recovery (FLAIR); sagittal T1 FLAIR at 9 months. b Axial 3D FLAIR; sagittal T1 at 21 months.
Developmental delay was present in 86% (12/14) of individuals. This included gross motor delays with delayed walking in 5/14 patients. Walking was achieved between 10 months and 5 years. Language delays included first words between 18 months and 4 years. Moreover, when speech was acquired, it is described as difficult to understand or dysarthric. Significant regression occurred in 6/14, following acute metabolic decompensation or major illness. Subsequently, some individuals regained skills. IQ ranged from normal to severe ID when testing was performed. Muscle biopsy demonstrated nonspecific myopathic changes in 3/4 patients. Neurologic exam findings included variable muscle tone abnormalities including hypotonia, hypertonia, dystonia, and upper motor neuron signs including hyperreflexia, clonus, and positive Babinski. Lower extremities were more significantly affected with spastic Achilles tendons. Two patients required surgical intervention and/or botulinum toxin injections. In three individuals, multiple joint contractures were present. Progressive microcephaly was seen in 5/14 patients.
Ophthalmologic findings
Ophthalmologic findings were variable and included optic nerve atrophy, cortical vision impairment in 2/14. Patient 1 had the most severe ophthalmologic findings with abnormal conjugate, volitional eye movements with nystagmus, and progressive optic nerve atrophy. Additionally, 3/14 had abnormal eye movements consisting of amblyopia and dysconjugate gaze.
Laboratory findings
Documented hypoglycemia occurred in 9/14 individuals during acute metabolic crisis or severe illness. Ammonia levels were elevated in 3/14 patients with lactic acidosis present during metabolic crisis in 8/14 patients. Creatine kinase elevation was present in 11/14 patients ranging from 14,000 to 278,000 U/L with rhabdomyolysis documented in 9/14 patients. Elevation of transaminases was seen in 11/14 patients and was a direct result of rhabdomyolysis. Abnormalities of thyroid-stimulating hormone (TSH) and/or free T4 occurred in 7/14 patients with 5 patients receiving treatment with L-thyroxine.
Gastrointestinal findings
Of 14 patients, 8 had feeding difficulties requiring gastrostomy tube (GT) placements. Dysmotility was seen in 6/14 individuals with delayed gastric emptying studies. One patient required GT feeds only in the setting of decompensation.
Cardiac findings
Electrocardiogram abnormalities occurred in 6/14 individuals in the setting of metabolic crisis, including prolonged QTc, torsades de pointes, ventricular tachycardia and/or fibrillation. Cardiac arrest was seen in three individuals, one of whom had an implantable cardioverter-defibrillator (ICD). It is important to note that patients with severe arrhythmias were ultimately refractive to antiarrhythmia medication. Echocardiogram abnormalities included left ventricular hypertrophy, dilated cardiomyopathy, prominent trabeculations, and decreased left ventricular function when ill.
Causes of death
Of 14 individuals, 5 were deceased. Ages of death ranged from 8 months to 9.8 years. The time from initial symptoms to death ranged from 4 months to 9 years. The most frequent cause of death was cardiac arrest due to arrhythmias, which occurred in 4/5 deceased individuals. Arrhythmias at the time of death included uncontrolled ventricular tachycardia, long QT, and torsades de pointes. Only one individual had an ICD placed (patient 4). For patient 4, despite an ICD and antiarrhythmic medication during a hospitalization at 7 years 9 months, she died in the setting of uncontrollable sustained ventricular arrhythmias that was not responsive to antiarrhythmic medication. Patient 13 had ventricular tachycardia following her second metabolic crisis. It was initially responsive to flecainide; however, following 2 months of treatment she developed medication-resistant tachycardia and torsades de pointes causing her death. Patient 6 presented with a metabolic crisis, cardiomyopathy, and heart failure at 27 months that resolved; however, during a subsequent metabolic crisis at 3 years 10 months, she died due to sudden cardiac death. Patient 5, her sister, did not experience arrhythmias aside from her first metabolic crisis where she had dilated cardiomyopathy and long QT. She does not have an ICD and is not on antiarrhythmic medication. Her most recent 24-h Holter and echocardiogram have been normal. In family 1, where no arrhythmias have been documented, patient 1 died following initiation of comfort care after a significant course of epileptic encephalopathy, progressive cerebral and optic nerve atrophy, and visceral hyperalgesia.
Full case descriptions are provided in the Supplemental Material.
Discussion
We report clinical and molecular results for 14 patients with TANGO2-related disease. Most of our patients experienced metabolic crises, rhabdomyolysis, seizures, and arrhythmias, which is consistent with previously described cases. However, we find that the initial presentation was more variable than previously reported, and some patients presented with primarily neurologic features. This suggests while the full TANGO2-related phenotype often emerges over time, many patients can present with initial features that are not suggestive of the final diagnosis. For example, patient 1 was initially thought to have an epileptic encephalopathy and then experienced neurodegeneration, while his brother (patient 2) presented with a milder encephalopathy with epilepsy; neither experienced rhabdomyolysis or life-threatening arrhythmias. Moreover, it is important to emphasize the heterogeneity in TANGO2 as a progressive neurodegenerative disorder. We presented patients of different ages and functional states who are at different stages of the disorder. Variable expressivity is seen in families 1 and 3. In our series, significant decline during crises is apparent followed by full recovery, partial recovery, or overall loss of function. Moreover, progressive brain atrophy apparent in early to late stages of brain atrophy was a feature in 5/14 individuals.
Delayed or postmortem diagnosis occurred in a number of individuals, highlighting the importance of reanalysis of apparently negative ES as novel genes are identified as well as consideration of novel genes with atypical presentations.5 Moreover, the need to incorporate CNV and exon level deletion/duplication analysis is crucial as the intragenic deletion was missed with standard analysis of next-generation sequencing data in patient 12. Our results further illustrate the molecular heterogeneity seen in patients with TANGO2-related disease. Five of seven pathogenic variants identified in our cohort are novel, including splice site, missense, and nonsense variants as well as a single-exon deletion (Fig. 4).

Variants from the 14 patients described are shown above the gene depiction with red bars for deletions and lines for single nucleotide variants (SNV), and pathogenic variants from prior studies are shown below (light blue bars for deletions and lines for SNV) based on TANGO2 transcript NM152906.6 (2623 bp).
An emerging theme in clinical genetics is that individuals can have two or more Mendelian disorders.6,7 If an individual’s clinical symptoms are not fully explained by their initial diagnosis, it is important to consider additional diagnoses. This was highlighted by patient 12, who had a known diagnosis of 22q11.2 deletion syndrome (22q11.2DS). Beyond what is expected in his primary diagnosis, he began experiencing episodic weakness and metabolic abnormalities. Given these new symptoms, further molecular analysis was sought, revealing the exon 3–9 deletion in TANGO2, which is within the region of recurrent microdeletions for 22q11.2DS. There were direct implications given his dual diagnosis because medical management is critically important during metabolic decompensation.
Molecular diagnosis of rare metabolic disorders can be challenging given their often extreme clinical and genetic heterogeneity. ES is a powerful diagnostic tool but is limited by current knowledge. Our cases illustrate the utility of ES data reanalysis because discovery of novel disease genes can diagnose previously unsolved cases. In addition, incorporating CNV analysis in ES data is imperative for TANGO2-related disorder. The potential for life-limiting medical complications such as metabolic crises and arrhythmias underscores the importance of early recognition and a rapid diagnosis of TANGO2-related disorder. The variability in initial presentation suggests that TANGO2 should be considered in the genetic workup of developmental delay, childhood-onset seizures, rhabdomyolysis, and suspected neurologic or metabolic disorders. Early diagnosis is important so that providers and families are aware of potential metabolic crises associated with illness. Further work is necessary, specifically relating to the function of TANGO2 and impact of specific variants on the function of the protein as this may lead to targeted therapies in affected individuals.
URLs
The URLs for data presented include:
gnomAD, http://gnomad.broadinstitute.org/
UCSC Genome Browser, https://genome.ucsc.edu/
ExAC Browser, http://exac.broadinstitute.org/
Change history
15 October 2018
The original version of this Article contained an error in the spelling of the author J. Lawrence Merritt, which was incorrectly given as Lawrence Merritt. This has now been corrected in both the PDF and HTML versions of the Article.
References
Kremer LS, Distelmaier F, Alhaddad B, et al. Bi-allelic truncating mutations in TANGO2 cause infancy-onset recurrent metabolic crises with encephalocardiomyopathy. Am J Hum Genet . 2016;98:358–362.
Lalani SR, Liu P, Rosenfeld JA, et al. Recurrent muscle weakness with rhabdomyolysis, metabolic crises, and cardiac arrhythmia due to bi-allelic TANGO2 mutations. Am J Hum Genet . 2016;98:347–357.
Retterer K, Scuffins J, Schmidt D, et al. Assessing copy number from exome sequencing and exome array CGH based on CNV spectrum in a large clinical cohort. Genet Med . 2015;17:623–629.
Kato T, Tamiya G, Koyama S, et al. UBR5 gene mutation is associated with familial adult myoclonic epilepsy in a Japanese family. ISRN Neurol. 2012;2012:508308.
Eldomery MK, Coban-Akdemir Z, Harel T, et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017;9:26.
Jehee FS, de Oliveira VT, Gurgel-Giannetti J, et al. Dual molecular diagnosis contributes to atypical Prader-Willi phenotype in monozygotic twins. Am J Med Genet A. 2017;173:2451–2455.
Posey JE, Harel T, Liu P, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. 2017;376:21–31.
Acknowledgements
We thank the families, GeneDx, and practitioners for their participation and collaboration in this case series. Research reported in this publication was supported in part by a grant from the National Institutes of Health (NS069605 to H.C.M.), and the National Institute of General Medical Sciences of the National Institutes of Health Postdoctoral Training Program in Medical Genetics (5T32GM007454 to J.N.D.). This work was also supported by intramural funds of the University of Kiel (I.H.), the German Research Foundation (HE5415/3-1 to I.H.) within the EuroEPINOMICS framework of the European Science Foundation, and individual grants by the German Research Foundation (D.F.G., HE5415/5-1, HE5415/6-1 to I.H.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
M.C., K.M., S.S., G.D., F.M., A.B., and K.G.M. are employees of GeneDx, a wholly owned subsidiary of OPKO Health. The other authors declare no conflicts of interest.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dines, J.N., Golden-Grant, K., LaCroix, A. et al. TANGO2: expanding the clinical phenotype and spectrum of pathogenic variants. Genet Med 21, 601–607 (2019). https://doi.org/10.1038/s41436-018-0137-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0137-y
Keywords
This article is cited by
-
Systematic reanalysis of genomic data by diagnostic laboratories: a scoping review of ethical, economic, legal and (psycho)social implications
European Journal of Human Genetics (2024)
-
Quality of life, illness perceptions, and parental lived experiences in TANGO2-related metabolic encephalopathy and arrhythmias
European Journal of Human Genetics (2022)
-
Mitochondrial dysfunction associated with TANGO2 deficiency
Scientific Reports (2022)
-
Genetic and Clinical Predictors of Ataxia in Pediatric Primary Mitochondrial Disorders
The Cerebellum (2022)
-
Integration of genetic, transcriptomic, and clinical data provides insight into 16p11.2 and 22q11.2 CNV genes
Genome Medicine (2021)
