
Abstract
Purpose
Genotyping CYP2D6 is important for precision drug therapy because the enzyme it encodes metabolizes approximately 25% of drugs, and its activity varies considerably among individuals. Genotype analysis of CYP2D6 is challenging due to its highly polymorphic nature. Over 100 haplotypes (star alleles) have been defined for CYP2D6, some involving a gene conversion with its nearby nonfunctional but highly homologous paralog CYP2D7. We present Stargazer, a new bioinformatics tool that uses next-generation sequencing (NGS) data to call star alleles for CYP2D6 (https://stargazer.gs.washington.edu/stargazerweb/). Stargazer is currently being extended for other pharmacogenes.
Methods
Stargazer identifies star alleles from NGS data by detecting single nucleotide variants, insertion-deletion variants, and structural variants. Stargazer detects structural variation, including gene deletions, duplications, and conversions, by calculating paralog-specific copy numbers from read depths.
Results
We applied Stargazer to the NGS data of 32 ethnically diverse HapMap trios that were genotyped by TaqMan assays, long-range polymerase chain reaction, quantitative multiplex polymerase chain reaction, high-resolution melting analysis, and/or Sanger sequencing. CYP2D6 genotyping by Stargazer was 99.0% concordant with the data obtained by these methods, and showed that 28.1% of the samples had structural variation including CYP2D6/CYP2D7 hybrids.
Conclusion
Accurate genotyping of pharmacogenes with NGS and subsequent allele calling with Stargazer will aid the implementation of precision drug therapy.
Similar content being viewed by others
INTRODUCTION
Many cytochrome P450 enzymes play a role in pharmacological responses by contributing to the metabolism of numerous drugs. Among these, cytochrome P450 2D6 (CYP2D6) is considered one of the most important because it contributes to the metabolism of about 25% of drugs.1 Drugs metabolized by CYP2D6 include opioids, chemotherapeutic agents, antidepressants, and antipsychotics, among others.2
The activity of CYP2D6 varies considerably between individuals due to the high level of polymorphisms observed in the CYP2D6 gene. There are more than 100 haplotypes defined for CYP2D6 by the Pharmacogene Variation Consortium.3 These are called star alleles (e.g., CYP2D6*1, *2, etc.) and are characterized by single nucleotide variants (SNVs), insertion-deletion variants (indels), structural variants (SVs), or a combination of these. They include fully functional, decreased-function, and nonfunctional alleles, which provide a wide spectrum of CYP2D6 enzymatic activity ranging from ultrarapid to poor metabolism. Different ethnic groups have distinct frequencies of star alleles and metabolic phenotypes;4 however, further studies are warranted for individuals of African or Asian ancestry because these populations are underrepresented in the estimation of CYP2D6 genetic diversity.5
Drug therapy without preemptive knowledge of a patient’s CYP2D6 phenotype status can lead to severe adverse reactions or a loss of efficacy due to inappropriate drug choice and/or dosing. For example, codeine is one of the most common and widely used opioids whose analgesic effect is elicited by CYP2D6 through the formation of morphine. Patients who are CYP2D6 poor metabolizers exhibit very low plasma concentrations of morphine following codeine administration, which can complicate their pain management because the affinity of morphine to the μ-opioid receptor is 200-fold stronger compared with that of codeine.6 Conversely, a patient can experience life-threatening morphine intoxication after receiving a small dose of codeine if they are a CYP2D6 ultrarapid metabolizer.7 Similarly, a breastfed infant died from morphine poisoning because the mother was prescribed a normal dose of codeine for childbirth-related pain. In this case, the mother was a CYP2D6 ultrarapid metabolizer passing a toxic amount of morphine to her newborn through her breast milk.8
For these reasons, there is considerable interest in genotyping CYP2D6. However, genotype analysis of CYP2D6 is complex because a large fraction of its existing variation cannot be accurately assessed with a single approach. SVs in CYP2D6, such as gene deletions, duplications, and conversions, are particularly challenging to detect due to high sequence homology (>95%) with a nonfunctional paralog, CYP2D7, located upstream of CYP2D6.9 Therefore, CYP2D6 is prone to genotype misclassification and incorrect phenotype prediction.10 In laboratory settings, several orthogonal genotyping methods, such as TaqMan assays, long-range polymerase chain reaction (PCR), quantitative multiplex PCR, high-resolution melting analysis, and Sanger sequencing, are employed to call star alleles. However, many of these methods are time consuming and heavily biased toward the detection of known variants. In clinical settings, due to practical limitations, only a handful of major star alleles, if any, are tested. Hence, a new approach for genotyping CYP2D6 is needed that is more robust and capable of higher throughput.
In this study, we developed Stargazer, a new bioinformatics tool for calling star alleles in CYP2D6 from next-generation sequencing (NGS) data. NGS is a powerful platform for variant detection because of its high-throughput data generation, comprehensive genotyping capabilities, and ever-decreasing cost. Additionally, NGS does not require previous knowledge about the variants of interest, and can uncover novel functional variants, which is not possible for many of the aforementioned genotyping methods. Furthermore, its cost-effectiveness can be increased for variant discovery by applying custom capture panels. To assess the accuracy of Stargazer, we applied it to the NGS data of 32 ethnically diverse HapMap trios. We report a correlation of 99.0% between CYP2D6 genotype calls determined with Stargazer and by orthogonal methods. We are now extending Stargazer to call star alleles for other clinically important pharmacogenes. Accurate diplotype calls from NGS data using Stargazer provide a promising approach for precision medicine to maximize drug efficacy and minimize toxicity for individual patients. Stargazer is publicly accessible through https://stargazer.gs.washington.edu/stargazerweb/.
MATERIALS AND METHODS
Samples
We built Stargazer using NGS data from 32 ethnically diverse trios. These trios were selected from the International HapMap Project, and they are comprised of 13 European, 5 Yoruban, 4 African American, 3 Han Chinese, 3 Mexican American, 2 Peruvian, and 2 Puerto Rican families (Table 1). These trios were originally sequenced to assess the performance of PGRNseq, a recently developed custom capture panel of key pharmacogenes including CYP2D6.11 They were specifically chosen for this study because they are a genetically diverse set of samples in which we would likely encounter a wide range of CYP2D6 variants, including SVs, to test Stargazer’s genotyping abilities and limitations. In addition, these trios allow for the analysis of Mendelian inheritance patterns to further the validation of Stargazer’s star allele calls. These trios were also previously genotyped for CYP2D6 by a variety of orthogonal methods (see below), allowing us to assess the accuracy of Stargazer’s diplotype calls.
Orthogonal genotyping methods
HapMap trios were genotyped for CYP2D6 according to procedures described elsewhere.12,13,14,15 Briefly, SNVs and indels were detected using TaqMan assays. Gene deletions, duplications, and multiplications were assessed by long-range PCR and quantitative multiplex PCR. CYP2D6/CYP2D7 hybrids were identified using quantitative multiplex PCR, high-resolution melting analysis, and/or Sanger sequencing.
Custom capture panel and NGS
HapMap trios were sequenced twice—once with PGRNseq v1.1 and once with PGRNseq v2.0—to a mean coverage of ~400× and ~160×, respectively. Both sequencing runs were performed with Illumina HiSeq 2500 machines using 100-base pair (pb) paired-end reads. Three samples failed during one of the two sequencing runs: NA19835 in PGRNseq v1.1, and NA19686 and NA11834 in PGRNseq v2.0. Note that the probes designed to capture CYP2D6 and CYP2D7 were more specific and extensive in PGRNseq v2.0 compared with PGRNseq v1.1; however, both versions generated reads that mapped to all the exons, introns, untranslated regions, and promoters of CYP2D6 and CYP2D7 (Supplementary Figure S1 online). Two samples, NA12878 and NA19238, were also previously sequenced by genome sequencing (WGS) to a mean coverage of ~30× with Illumina HiSeq X instruments using 150-bp paired-end reads. These data were used to test Stargazer’s generalizability to WGS data.
Input and output data of Stargazer
The Stargazer CYP2D6 genotyping pipeline is outlined in Figure 1. The pipeline uses BAM files comprising sequence reads aligned with BWA-MEM to human reference genome assembly GRCh37.16 BAM files are then used to generate a VCF file with GATK-HaplotypeCaller (v3.4),17 from which Stargazer extracts all SNVs and indels located within 3 kilobases (kb) from either end of CYP2D6. More specifically, Stargazer stores the genomic position of each variant, reference allele, alternate allele(s), genotype status (homozygous or heterozygous), and allelic depth for each sample. Stargazer uses the variant information from the VCF file to call star alleles based on SNVs and indels.

Stargazer takes as input a VCF file, target GDF file, and control GDF file. It uses the variant information from the VCF file to call star alleles based on SNVs and indels. Using the target and control GDF files, Stargazer converts read depth to copy number for detection of structural variation. The output data of Stargazer include each sample’s CYP2D6 diplotype and plots to visually inspect copy number for CYP2D6 and CYP2D7. Based on called CYP2D6 diplotypes, the program outputs predicted phenotypes as well. Several external software tools, shown in red, are used both within and outside Stargazer
BAM files are also used to calculate read depth for CYP2D6 and CYP2D7 with GATK-DepthOfCoverage (v3.4).17 For convenience, we will refer to this output as a target GDF (GATK-DepthOfCoverage format) file. Since the high homology between CYP2D6 and CYP2D7 can cause reads to align to erroneous or multiple locations, only uniquely mapping reads with a mapping quality ≥20 are counted. Similarly, a control GDF file is produced from a user-chosen locus, which serves as a read depth normalization factor. Stargazer computes paralog-specific copy numbers using read depth from the target and control GDF files in order to detect SVs.
In the initial development of Stargazer, three genes—VDR, RYR1, and EGFR—were evaluated as control loci. These genes are covered by PGRNseq and are 63, 154, and 188 kb in size, respectively. They are also reported to exhibit low rates of whole gene deletion and/or duplication according to the Database of Genomic Variants.18 All three genes produced the same copy number results for CYP2D6 and CYP2D7. The analyses shown in the results section were all performed using RYR1 as the control locus.
The output data of Stargazer include each sample’s CYP2D6 diplotype, predicted phenotype, and plots to visually inspect copy number for CYP2D6 and CYP2D7 (Figure 2). Note that when calling diplotypes, Stargazer only considers those variants that are currently used by the Pharmacogene Variation Consortium. Stargazer also returns all detected SNVs and indels, including those that are novel and those that are known but not currently used to define any star allele. As follow-up, these variants can be functionally annotated using variant annotation tools such as SeattleSeq Annotation (http://snp.gs.washington.edu/SeattleSeqAnnotation).

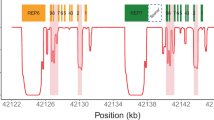
a European sample NA12805, which has a CYP2D6*2/*4 diplotype without structural variation, was included for comparison. b Gene deletion in Yoruban sample NA18508 with a CYP2D6*2/*5 diplotype. c Gene duplication in Mexican American sample NA19685 with a CYP2D6*1/*2 × 2 diplotype. d Complex structural variation involving a gene duplication and a gene conversion in Peruvian sample HG01979 genotyped as CYP2D6*2/*68 + *4. e Complex structural variation involving multiple gene duplications and gene conversions in Han Chinese sample HG00465 genotyped as CYP2D6*36 + *10/*36 + *10. f Complex structural variation involving a gene conversion in Mexican American sample NA19790 genotyped as CYP2D6*1/*78 + *2. Gray dots show the copy number calculated from the read depth. Dots colored purple and orange represent the mean copy numbers for CYP2D6 and CYP2D7, respectively, as determined by the changepoint algorithm. Each panel contains scaled CYP2D6 and CYP2D7 gene models, in which the exons and introns are depicted using boxes and lines, respectively. All panels were generated from PGRNseq v2.0 data
Prediction of star alleles
From a VCF file, Stargazer uses Beagle (v4.1)19 to haplotype phase heterozygous variants for CYP2D6 with over 2,500 reference samples from the 1000 Genomes Project. Stargazer then matches phased haplotypes to star alleles using a translation table built from publicly available data (https://www.pharmvar.org). The table contains information on more than 90 star alleles and 185 SNVs and indels, including variant positions and nucleotide changes in relation to the reference CYP2D6*1 allele and human reference genome assembly GRCh37.
Detection of SVs
From a target GDF file, Stargazer converts read depths for CYP2D6 and CYP2D7 to copy numbers by performing intra- and intersample normalizations. Intrasample normalization accounts for individual variation in the sequencing efficiency using read depth from a control GDF file, while the intersample normalization considers the heterogeneity in coverage across all samples. Stargazer then automates the detection of SVs with changepoint (v2.2.2)—an R package that approximates one or more points at which the statistical properties of a sequence of observations change.20 Here, the sequence is DNA, the observation is per-base copy number, and the statistical property is the mean copy number. If there is a significant shift in the mean copy number (e.g., from two to one), the algorithm returns the change point location and the two mean values (e.g., two and one).
Identification of diplotypes
For samples without SVs, Stargazer determines CYP2D6 diplotypes by combining the star allele used to assign each phased haplotype. For samples with a whole gene deletion, the affected haplotype is assigned the CYP2D6*5 deletion allele, which is then combined with the star allele assigned to the other haplotype to form a diplotype. For samples with a whole gene duplication, the affected haplotype is assigned “×2” (e.g., CYP2D6*1 × 2, *2 × 2, etc.) because it has two gene copies of CYP2D6. For samples with more complex SVs, such as CYP2D6/CYP2D7 hybrids, individual algorithms have been developed to determine diplotypes. The identification of diplotypes is discussed in more detail, in the context of HapMap trios, in the results section.
Assignment of predicted phenotypes
There are four CYP2D6 metabolizer classes: poor, intermediate, normal, and ultrarapid. To predict these phenotypes, Stargazer first translates CYP2D6 diplotypes into a standard unit of enzyme activity known as an activity score.21 The fully functional reference CYP2D6*1 allele is assigned a value of 1, decreased-function alleles such as CYP2D6*10 and *17 receive a value of 0.5, and nonfunctional alleles including CYP2D6*4 and *5 have a value of 0. The sum of values assigned to both alleles constitutes the activity score of a diplotype. Consequently, subjects with CYP2D6*1/*1, *1/*4, and *4/*5 diplotypes have an activity score of 2, 1, and 0, respectively. These activity scores are used to predict the four metabolizer classes as follows: poor, 0; intermediate, 0.5; normal, 1–2; and ultrarapid, >2.
RESULTS
Identification of diplotypes by Stargazer for HapMap trios
We used Stargazer (v1.0.0) to call CYP2D6 diplotypes for 32 HapMap trios sequenced with PGRNseq (Table 1). Data from PGRNseq v1.1 and PGRNseq v2.0 served as technical validation and produced the same diplotype calls for all samples. Moreover, all diplotype calls were inherited in a predictable manner, as exemplified in Figure 3. Diplotypes were identified using the following algorithms.

a Segregation of CYP2D6*78 + *2 in the Mexican American M037 family. b Segregation of CYP2D6*68 + *4 in the European 1463 family. The top, middle, and bottom rows show data from the father, mother, and child, respectively. Gray dots show the copy number calculated from the read depth. Dots colored purple and orange represent the mean copy numbers for CYP2D6 and CYP2D7, respectively, as determined by the changepoint algorithm. Each panel contains scaled CYP2D6 and CYP2D7 gene models, in which the exons and introns are depicted using boxes and lines, respectively. All panels were generated from PGRNseq v2.0 data
For samples without SVs, diplotypes were determined by combining the star allele assigned to each of the two haplotypes. For example, phasing algorithms estimated from subject NA12805 two haplotypes—of which one matched CYP2D6*2 and the other *4—to form a CYP2D6*2/*4 diplotype (Figure 2a).
For samples with a whole gene deletion, diplotypes were determined such that one haplotype contained the CYP2D6*5 deletion allele while the other was assigned a star allele based on detected SNVs and indels. For example, subject NA18508 had only one CYP2D6 gene copy, and all detected SNVs were hemizygous and matched CYP2D6*2. Stargazer called this sample as having a CYP2D6*2/*5 diplotype (Figure 2b).
For samples with a whole gene duplication, Stargazer resolved the identity of the extra CYP2D6 gene copy in the affected haplotypes. For example, Stargazer detected three gene copies in subject NA19685 with a CYP2D6*1/*2 diplotype (Figure 2c). This sample could tentatively have a duplication on the CYP2D6*1 or *2 allele, or in other words could have a CYP2D6*1 × 2/*2 or *1/*2 × 2 diplotype. Stargazer used the allelic depth ratios of the SNVs defining the CYP2D6*2 allele to determine which allele carried the duplication. If the CYP2D6*2 allele carried the extra copy, the sample would have a read ratio of 2:1 reads for the respective SNVs. Indeed, most samples that were heterozygous for these SNVs had read ratios close to 1, whereas NA19685 was a significant outlier with an average ratio of 2.4 from the PGRNseq 1.1 data. Stargazer called the sample as having a CYP2D6*1/*2 × 2. The read ratio approach can also be used to distinguish between diplotypes having duplications on both alleles and those having multiple copies on one allele (e.g., CYP2D6*1 × 2/*2 × 2 vs. *1/*2 × 3).
For samples with complex structural variation, diplotypes were called using individual algorithms. For example, CYP2D6*68 + *4 is a tandem duplication where the CYP2D6*4 gene copy is defined by only one SNV while CYP2D6*68 is a hybrid gene featuring a CYP2D7 sequence from intron 1 onward and four CYP2D6 SNVs before the breakpoint. Stargazer called one of the two haplotypes of HG01979 as having this tandem structure because all five SNVs were detected, they were haplotype phased together, and the conversion to CYP2D7 was also observed (Figure 2d). The other haplotype was matched to CYP2D6*2 and Stargazer called this sample as having a CYP2D6*2/*68 + *4 diplotype. Similar approaches were employed to determine diplotypes involving other tandem duplications, such as CYP2D6*36 + *10 (Figure 2e) and *78 + *2 (Figure 2f), where the CYP2D6*36 and *78 alleles each contain a gene conversion to CYP2D7.
For samples with more than one SV, Stargazer tested all possible pairwise combinations of SVs to determine diplotypes. More specifically, Stargazer first fit every combination of SVs against a sample’s observed CYP2D6 and CYP2D7 copy number profiles and then selected the combination that produced the least deviance. For example, HG00463 and HG00465 (family SH021) carry both a gene duplication and a gene conversion on each of their chromosomes, and their profiles are best explained as having a CYP2D6*36 + *10 tandem arrangement on each chromosome (Supplementary Figure S2a,b online). There was one additional sample with multiple SVs—HG01190—whose profile was best explained by the combination of a CYP2D6*5 deletion and CYP2D6*68 + *4 (Supplementary Figure S2c).
Summary of genotype and phenotype calls by Stargazer for HapMap trios
Stargazer called 20 unique haplotypes from the 32 HapMap trios. The frequencies of these haplotypes in the 64 unrelated parents are shown in Supplementary Table S1 online. As expected, fully functional CYP2D6*1 and *2 alleles had the highest frequencies (31.3 and 18.8%, respectively) among the parents, followed by the nonfunctional CYP2D6*4 allele (8.6%). Stargazer also detected CYP2D6*17 (7.0%), which is commonly found in subjects of African ancestry including African Americans, and CYP2D6*10 (2.3%), which occurs most frequently in East Asians. Both CYP2D6*17 and *10 are decreased-function alleles. In addition, Stargazer identified many haplotypes with structural variation in the parents: 4.7% with a gene deletion (CYP2D6*5), 11.7% with a gene duplication (CYP2D6*2 × 2, *4 × 2, *4 N + *4, *10 × 2, *36 + *10, *68 + *4, and *78 + *2), and 8.6% with a gene conversion (CYP2D6*4 N, *36, *68, and *78). This translates to 9.4, 21.9, and 15.6% of the parents having at least one gene deletion, duplication, or conversion, respectively. Based on the diplotype calls, 4.7, 10.9, 82.8, and 1.6% of the parents were predicted to be poor, intermediate, normal, and ultrarapid metabolizers, respectively.
Comparison between Stargazer and orthogonal genotyping methods
When the CYP2D6 haplotype calls of Stargazer were compared with those determined by orthogonal methods, the concordance rate was 99.0% (190 out of 192 haplotypes; Table 1). The two discordant haplotypes were found in NA19200 and NA19202 from the Y045 trio. For these samples, the orthogonal methods called CYP2D6*5/*76 + *1 and CYP2D6*1/*76 + *1 diplotypes, respectively, while Stargazer called CYP2D6*1/*5 and CYP2D6*1/*1 diplotypes. The nonfunctional CYP2D6*76 allele—a CYP2D6/CYP2D7 hybrid—was identified using long-range PCR and Sanger sequencing. The allele is essentially a CYP2D7 gene that has a CYP2D6 downstream sequence with a switch region to CYP2D6 past exon 9, and lacks a CYP2D7-specific sequence also referred to as ‘spacer.’22 Since this allele has a CYP2D6-specific sequence, it may produce positive results with some long-range PCR reactions that are deemed diagnostic for the presence of a gene duplication. Nonetheless, Stargazer did not detect any significant change in copy number either at the switch region or in the spacer, so the program did not call CYP2D6*76 (Supplementary Figure S3 online). Note that the phenotype prediction is the same whether the allele is detected or not.
Stargazer calls using WGS data
To assess the performance of Stargazer on WGS data, we evaluated two samples (NA19238 and NA12878) that were sequenced with PGRNseq v1.1, PGRNseq v2.0, and WGS. Although NA12878 carried a CYP2D6*68 + *4 tandem duplication, Stargazer called the same diplotypes regardless of the sequencing platform (Figure 4).

a–c Two subjects, NA19238 and NA12878, were sequenced with a PGRNseq v1.1 at ~400× coverage with 100-base pair (bp) paired-end reads, b PGRNseq v2.0 at ~160× coverage with 100-bp paired-end reads, and c genome sequencing at ~30× coverage with 150-bp paired-end reads. Gray dots show the copy number calculated from the read depth. Dots colored purple and orange represent the mean copy numbers for CYP2D6 and CYP2D7, respectively, as determined by the changepoint algorithm. Each panel contains scaled CYP2D6 and CYP2D7 gene models, in which the exons and introns are depicted using boxes and lines, respectively. In all three cases, Stargazer called the correct diplotypes: CYP2D6*1/*17 and *3/*68 + *4, respectively
Testing Stargazer at various sequencing coverages
We applied Stargazer to simulated datasets generated by randomly downsampling sequence reads from PGRNseq v2.0 data (Supplementary Table S2 online). When 15% of reads were used (corresponding to 23.7× coverage), 186 out of 188 haplotypes were correctly called; the two misclassified haplotypes carried an SV. However, the misclassifications did not affect the phenotype prediction. Based on these results, our recommendation is to use Stargazer for datasets with a mean read coverage greater than 20×.
SNVs and indels detected by NGS
From the PGRNseq v1.1 and PGRNseq v2.0 data, 142 SNVs and indels were detected at 138 loci within 3 kb from either end of CYP2D6, 86 of which are not currently used to define star alleles (Supplementary Table S3 online). Among these—according to SeattleSeq Annotation—five are missense pathogenic variants while the remaining ones are either synonymous or within the 3′ or 5′ untranslated region, downstream or upstream of the gene, or within an intron. We did not find any novel variants that are obviously detrimental to CYP2D6 function, such as nonsense, frameshift, or splice site pathogenic variants.
DISCUSSION
We developed Stargazer—a new software tool for calling star alleles in various polymorphic pharmacogenes from NGS data. When building Stargazer, we used CYP2D6 as a model for the detection and interpretation of SVs in the context of other observed SNVs and indels. We purposefully chose CYP2D6 as a starting point because it is one of the most complex genetic loci to genotype in the human genome. Two other programs—Cypiripi and Astrolabe—have been published to genotype CYP2D6 from NGS data.23, 24 Although both tools can reliably call simple diplotypes, they have difficulties with the detection of complex SVs, such as CYP2D6/CYP2D7 hybrids. We show that Stargazer can reliably detect those hybrids from targeted or WGS data.
More specifically, we show that Stargazer correctly genotyped CYP2D6 for 32 ethnically diverse HapMap trios. These trios were previously validated by a variety of orthogonal methods, and comparisons show that Stargazer is 99.0% concordant with these methods. In the future, we will test additional verified samples in order to further validate Stargazer’s performance. All diplotype calls by Stargazer were inherited according to expectations including population-specific star alleles such as CYP2D6*10 and *17. Stargazer also produced the same diplotype calls for all samples from the two independent PGRNseq v1.1 and PGRNseq v2.0 datasets.
We plan to extend Stargazer to CYP2A6—another highly polymorphic pharmacogene displaying many SNVs and indels as well as SVs. CYP2A6 metabolizes nicotine, and sequence variation in CYP2A6 has been linked to nicotine dependence and withdrawal symptoms upon smoking cessation.25 Similar to CYP2D6, CYP2A6 has several star alleles with a gene conversion to its nearby paralog CYP2A7.26 We also plan to develop Stargazer for other cytochrome P450 genes.
As larger genomic datasets become available, several aspects of Stargazer will improve. These include the statistical estimation of phased haplotypes, primarily haplotypes based on rare variants. In the current version of Stargazer, we incorporated a large panel of reference samples from the 1000 Genomes Project. This approach performed well for our dataset, but we are aware that in further applications, rare variants may have frequencies that are too low to be phased reliably. To ameliorate this issue, we plan to merge multiple large reference panels to obtain additional haplotype information. Novel variants will require physical phasing backed by sequence reads. When short reads cannot provide adequate phasing, long-read sequencing from Oxford Nanopore Technologies or Pacific Biosciences can be used to generate reference haplotype information. Recently, both technologies have been successfully applied to sequence CYP2D6.27, 28
Certain features of Stargazer are specific for targeted sequencing such as PGRNseq. For example, for the purpose of normalization, Stargazer requires multiple samples to be analyzed at a time. This is because sequencing with custom capture typically yields uneven coverage across the genes of interest, and Stargazer’s copy number estimation is based on population statistics. If the sample size is too small or a large fraction of samples share the same type of structural variation, population statistics can be shifted dramatically, generating biased copy number data. However, this problem can be addressed by including reference samples with known copy numbers. For WGS data, where coverage is usually distributed more evenly, the intersample normalization may be skipped, allowing Stargazer to analyze a single sample.
We reported five missense variants that are not currently used to define any star allele. However, interpretation of these variants is difficult without functional characterization. In fact, the same is true for many variants in existing star alleles (e.g., CYP2D6*22 is defined by a nonsynonymous SNV in exon 9 with unknown effect). Therefore, there is clearly a need to more rigorously characterize the function of the rapidly increasing number of haplotypes to facilitate phenotype prediction. In the future, it is possible that data from deep mutational scanning for the CYP2D6 enzyme could be incorporated into Stargazer to aid the characterization of the functional consequences of all possible single pathogenic variants of this protein.29
The HapMap trios used in this study consist of seven distinct ethnic groups and therefore represent a sampling of the global distribution of CYP2D6 genotypes. Characterized by multiple genotyping platforms including NGS, these trios can serve as a reference resource for other CYP2D6 genotyping projects.
There is growing awareness of individual variation in drug response. For example, in March 2013, the Food and Drug Administration cautioned against the use of codeine in children of any age to treat pain after surgery to remove the tonsils or adenoids.30 Shortly after, a prospective study showed that children who were CYP2D6 ultrarapid metabolizers and taking codeine after those surgeries were at a higher risk for toxicity and death.31 In April 2017, the Food and Drug Administration issued the agency’s strongest warning against codeine, alerting that the medication should not be used to treat pain or cough in children younger than 12 years. While limiting the therapeutic use of codeine addresses the concern for patient safety, tailoring codeine or other drug treatments based on an individual’s CYP2D6 genotype could achieve the same goal. There may also be therapeutic settings where alternative treatments are not fully interchangeable and health outcomes could suffer from restrictions in drug choice. For example, national guidelines recommend codeine as a front-line drug for the treatment of pain in patients with sickle cell disease, and many hematologists prefer codeine to other analgesics that have comparable efficacy but higher potential for abuse and physical dependence.32 With additional validation, Stargazer may offer an alternative approach for optimizing treatment response in all patients.
References
Zhou SF. Polymorphism of human cytochrome P450 2D6 and its clinical significance: part I. Clin Pharmacokinet. 2009;48:689–723.
Crews KR, Gaedigk A, Dunnenberger HM, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450 2D6 genotype and codeine therapy: 2014 update. Clin Pharmacol Ther. 2014;95:376–82.
Gaedigk A, Ingelman-Sundberg M, Miller NA, et al. The Pharmacogene Variation (PharmVar) Consortium: incorporation of the Human Cytochrome P450 (CYP) Allele Nomenclature Database. Clin Pharmacol Ther. 2018;103:399–401.
Gaedigk A, Sangkuhl K, Whirl-Carrillo M, Klein T, Leeder JS. Prediction of CYP2D6 phenotype from genotype across world populations. Genet Med. 2017;19:69–76.
LLerena A, Naranjo ME, Rodrigues-Soares F, Penas-LLedó EM, Fariñas H, Tarazona-Santos E. Interethnic variability of CYP2D6 alleles and of predicted and measured metabolic phenotypes across world populations. Expert Opin Drug Metab Toxicol. 2014;10:1569–83.
Kirchheiner J, Schmidt H, Tzvetkov M, et al. Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. Pharm J. 2007;7:257–65.
Gasche Y, Daali Y, Fathi M, et al. Codeine intoxication associated with ultrarapid CYP2D6 metabolism. N Engl J Med. 2004;351:2827–31.
Koren G, Cairns J, Chitayat D, Gaedigk A, Leeder SJ. Pharmacogenetics of morphine poisoning in a breastfed neonate of a codeine-prescribed mother. Lancet. 2006;368:704.
Gaedigk A. Complexities of CYP2D6 gene analysis and interpretation. Int Rev Psychiatry. 2013;25:534–53.
Naranjo ME, de Andrés F, Delgado A, Cobaleda J, Peñas-Lledó EM, LLerena A. High frequency of CYP2D6 ultrarapid metabolizers in Spain: controversy about their misclassification in worldwide population studies. Pharm J. 2016;16:485–90.
Gordon AS, Fulton RS, Qin X, Mardis ER, Nickerson DA, Scherer S. PGRNseq: a targeted capture sequencing panel for pharmacogenetic research and implementation. Pharm Genom. 2016;26:161–8.
Gaedigk A, Bradford LD, Alander SW, Leeder JS. CYP2D6*36 gene arrangements within the CYP2D6 locus: association of CYP2D6*36 with poor metabolizer status. Drug Metab Dispos. 2006;34:563–9.
Gaedigk A, Ndjountché L, Divakaran K, et al. Cytochrome P4502D6 (CYP2D6) gene locus heterogeneity: characterization of gene duplication events. Clin Pharmacol Ther. 2007;81:242–51.
Gaedigk A, Fuhr U, Johnson C, Bérard LA, Bradford D, Leeder JS. CYP2D7–2D6 hybrid tandems: identification of novel CYP2D6 duplication arrangements and implications for phenotype prediction. Pharmacogenomics. 2010;11:43–53.
Gaedigk A, Twist GP, Leeder JS. CYP2D6, SULT1A1 and UGT2B17 copy number variation: quantitative detection by multiplex PCR. Pharmacogenomics. 2012;13:91–111.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–60.
McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42:D986–92.
Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–97.
Killick R, Eckley IA. changepoint: an R package for changepoint analysis. J Stat Softw. 2014;58:1–19.
Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharmacol Ther. 2008;83:234–42.
Gaedigk A, Jaime LK, Bertino JS, et al. Identification of novel CYP2D7–2D6 hybrids: non-functional and functional variants. Front Pharmacol. 2010;1:121.
Numanagić I, Malikić S, Pratt VM, Skaar TC, Flockhart DA, Sahinalp SC. Cypiripi: exact genotyping of CYP2D6 using high-throughput sequencing data. Bioinformatics. 2015;31:i27–34.
Twist GP, Gaedigk A, Miller NA, et al. Constellation: a tool for rapid, automated phenotype assignment of a highly polymorphic pharmacogene, CYP2D6, from whole-genome sequences. NPJ Genom Med. 2016;1:15007.
Kubota T, Nakajima-Taniguchi C, Fukuda T, et al. CYP2A6 polymorphisms are associated with nicotine dependence and influence withdrawal symptoms in smoking cessation. Pharm J. 2006;6:115–9.
Fukami T, Nakajima M, Sakai H, McLeod HL, Yokoi T. CYP2A7 polymorphic alleles confound the genotyping of CYP2A6*4A allele. Pharm J. 2006;6:401–12.
Ammar R, Paton TA, Torti D, Shlien A, Bader GD. Long read nanopore sequencing for detection of HLA and CYP2D6 variants and haplotypes. F1000Res. 2015;4:17.
Qiao W, Yang Y, Sebra R, et al. Long-read single molecule real-time full gene sequencing of cytochrome P450-2D6. Hum Mutat. 2016;37:315–23.
Fowler DM, Fields S. Deep mutational scanning: a new style of protein science. Nat Methods. 2014;11:801–7.
Kuehn BM. FDA: no codeine after tonsillectomy for children. JAMA. 2013;309:1100.
Prows CA, Zhang X, Huth MM, et al. Codeine-related adverse drug reactions in children following tonsillectomy: a prospective study. Laryngoscope. 2014;124:1242–50.
Gammal RS, Crews KR, Haidar CE, et al. Pharmacogenetics for safe codeine use in sickle cell disease. Pediatrics 2016 Jul;138. pii: e20153479.
ACKNOWLEDGMENTS
The authors acknowledge the Pharmacogenomics Research Network for supporting the development of PGRNseq. This work was supported by NIH grants HL069757, GM092676, GM116691, GM115318, GM115277, and S10OD021553, and the University of Washington’s Graduate School Fund for Excellence and Innovation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Lee, Sb., Wheeler, M.M., Patterson, K. et al. Stargazer: a software tool for calling star alleles from next-generation sequencing data using CYP2D6 as a model. Genet Med 21, 361–372 (2019). https://doi.org/10.1038/s41436-018-0054-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0054-0
Keywords
This article is cited by
-
Mining local exome and HLA data to characterize pharmacogenetic variants in Saudi Arabia
Human Genetics (2024)
-
Pharmacovariome scanning using whole pharmacogene resequencing coupled with deep computational analysis and machine learning for clinical pharmacogenomics
Human Genomics (2023)
-
Genome screening, reporting, and genetic counseling for healthy populations
Human Genetics (2023)
-
Development of an extensive workflow for comprehensive clinical pharmacogenomic profiling: lessons from a pilot study on 100 whole exome sequencing data
The Pharmacogenomics Journal (2022)
-
Lack of association of CYP2B6 pharmacogenetics with cyclophosphamide toxicity in patients with cancer
Supportive Care in Cancer (2022)
