
Abstract
The innate immune response is the major front line of defense against viral infections. It involves hundreds of genes with antiviral properties which expression is induced by type I interferons (IFNs) and are therefore called interferon stimulated genes (ISGs). Type I IFNs are produced after viral recognition by pathogen recognition receptors, which trigger a cascade of activation events. Human and mouse studies have shown that defective type I IFNs induction may hamper the ability to control viral infections. In humans, moderate to high-effect variants have been identified in individuals with particularly severe complications following viral infection. In mice, functional studies using knock-out alleles have revealed the specific role of most genes of the IFN pathway. Here, we review the role of the molecular partners of the type I IFNs induction pathway and their implication in the control of viral infections and of their complications.
Similar content being viewed by others

Introduction
Interferons (IFNs) are cytokines that represent one of the first innate immune barriers against viruses. They were discovered in 1957 and were named after their capacity to “interfere” with virus replication. Recognition of non-specific viral molecules such as viral proteins, DNA, and RNA leads to their expression. After recognition of virus components by pathogen recognition receptors (PRR), an induction cascade leads to the activation of interferon regulatory factors (IRFs), the transcriptional factors responsible for IFN genes expression [1]. IFNs are glycoproteins that are secreted into the extracellular medium and act as autocrine and paracrine factors. The binding to their receptors induces the expression of interferon stimulated genes (ISGs) with antiviral properties. Non exhaustively, ISGs can inhibit nuclear import of nucleic acids, synthesis of RNA and proteins, or can enhance virus degradation [1]. Several proteins involved in IFN production and response are inhibited by nonstructural proteins of various viruses, which therefore escape host innate defense [2].
IFNs are grouped in three types depending on their sequence, structure, and function. IFNγ is the only type II IFN. It is produced by natural killer cells and binds the IFNγ receptor (IFNGR) composed of two subunits (IFNGR1/IFNGR2). This receptor recruits the Janus kinases 1 (JAK1) and 2 (JAK2), which activate the signal transducer and activator of transcription 1 (STAT1). STAT1 acts as homodimers [3] and binds gamma-activated sites present in the target ISGs promoters [1]. Type III IFNs include four IFN lambda numbered IFNλ1 to IFNλ4. The receptor to type III IFNs is composed of interleukin 28 receptor subunit alpha (IL-28Ra) and interleukin 10 receptor subunit 2 (IL-10R2). It induces the activation of the interferon stimulated gene factor 3 (ISGF3), composed of STAT1, STAT2 and IRF9, that binds to IFN-stimulated response elements on the promoter of target ISGs [4]. This review focuses on type I IFNs (IFN-I), which are among the first cytokines produced after viral infection [3]. IFN-I usually refer to IFNα and IFNβ, but also include other cell- and species-specific molecules. All IFN-I signal through the IFNα receptor composed of two subunits (IFNAR1 and IFNAR2), which recruit JAK1 and non-receptor tyrosine-protein kinase (TYK2). These kinases activate ISGF3 which binds to IFN-stimulated response elements [3].
Considering the crucial role of IFN-I in host responses to invading viruses, the inability to induce their expression often leads to severe symptoms. The variable outcome of viral infections has triggered genetic studies in humans and in mice [5, 6]. Unsurprisingly, genetic variants or deficiencies in IFN-I induction pathway genes were associated with susceptibility to diverse viruses. Here we provide a general presentation of the partners of this pathway, and we review the genetic susceptibilities to viral infections associated with these genes.
Molecular mechanisms of type I IFN induction
Type I interferons
IFN-I is the largest family of IFN proteins. They have a common helical structure composed of 5 α-helices and are encoded by genes clustered on chromosome 9 in humans and on chromosome 4 in mice [7].
The two main IFN-I are IFNα and IFNβ. These proteins are not constitutively expressed but are up-regulated during viral infection following the activation of the transcription factors IRF3 and IRF7 [8]. Most animal species have multiple IFNα genes, 13 genes with 80% nucleotide identity in human and 14 genes in mice. IFNαs are produced by plasmacytoid dendritic cells and hematopoietic cells (mostly leucocytes). Each type of IFNα has a different affinity for its receptor, and thus may trigger type-specific responses [9]. IFNβ is encoded by a single gene, IFNB1, and is also present in most animal species. It is produced by fibroblasts, dendritic cells, and epithelial cells [9].
Other IFNs-I have been described in animal species or in humans. Each of them is encoded by a single gene [9]. IFNε is constitutively expressed in the brain, lungs, small intestine and reproductive tissues. It is regulated by hormones and not during infections. IFNκ is present in a few species including humans and mice. It is constitutively expressed in keratinocytes and can be up-regulated after exposure to double-stranded RNA (dsRNA). IFNω is present in humans but not in mice. It is expressed mainly in leukocytes. IFNζ, also called limitin, is an IFN-like molecule present only in mice. It is expressed in mature T lymphocytes, bronchial, epithelial and salivary duct cells. IFNτ and IFNδ have been described but are not expressed in humans or in mice.
Virus recognition by PRR
The production of IFNβ and IFNα is induced by PRRs that recognize molecules present in pathogens called pathogen-associated molecular patterns. PRRs include Toll-like receptors (TLRs) and RIG-I-like receptors (RLRs). PRRs recognize components from bacteria, viruses and fungi and have specific ligands. During viral infections, TLR3 recognizes dsRNA while TLR7 and TLR8 recognize single-stranded RNA (ssRNA) and TLR9 recognizes DNA molecules. These TLRs are produced in the endoplasmic reticulum and sense their ligands in endosomes after virus entry into host cells [10]. TLR2 and TLR4 are present at the cell surface and recognize viral proteins [11, 12]. Replication of viruses with positive ssRNA genome produces dsRNA, which is recognized by TLR3 and RLRs [10].
Three RLRs recognize viral RNA. While DDX58 (also known as RIG-I) senses 5′-phosphorylated RNA, IFIH1 (MDA5) recognizes long dsRNA. DHX58 (LGP2) facilitates viral RNA recognition by DDX58 and IFIH1 [13] and enhances RLR-dependent IFN induction [14].
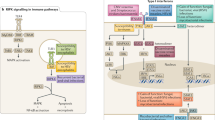
Viral recognition by TLRs and RLRs triggers a cascade of molecular activations, which results in the production of IFN-I. This pathway is summarized in Fig. 1.

Viral molecules (DNA, RNA and proteins) induce the expression of IFN-I after their recognition by TLRs and RLRs. Signalization leads the activation of kinases, TBK1 and IKKε responsible for the activation of the transcription factors IRF3 and IRF7 which induce the expression of IFNα and IFNβ. Proteins for which the corresponding gene was associated with susceptibility to virus infection are indicated in orange for mouse studies and in purple for human studies. Blue, yellow and green helices depict viral RNA, viral DNA and cellular DNA molecules, respectively. Gene names are spelled according to the nomenclature rules for human genes.
TLR pathway
TLR3 recognition of viral RNA induces its own phosphorylation, which allows the recruitment of the adaptor protein Toll-interleukin receptor (TIR) domain-containing adapter molecule 1 (TICAM1, also called TRIF) [15]. Interaction between TLR3 and TICAM1 is enabled by the phosphorylation of two TLR3 tyrosine residues [15]. TLR4 also can induce the expression of IFN-I by recognizing viral proteins present in the extracellular medium and signalling through the adaptors myeloid differentiation primary response protein (MYD88) and myelin and lymphocyte protein (MAL). Once activated, TLR4 is endocytosed and recruits TICAM1 and TIR domain-containing adapter molecule 2 (TICAM2, also called TRAM) in the endosomes [16]. TICAM1 recruits the TNF receptor associated factor 3 (TRAF3) [15], which then activates the kinases responsible for the activation of the IRFs. TICAM1 is targeted by the viral 3C protease of hepatitis A virus and coxsackievirus B3 (CVB3), which allows these viruses to escape the host immune response [10].
TLR2 also activates the expression of IFN-I, but the mechanisms are incompletely understood. Signalling by TLR2 requires MAL, TICAM2 and MYD88 which, once activated, relocate to the endosomes and induce a signalling cascade resulting in IRF7 activation and IFN-I expression. Therefore TLR2 and TLR4 likely use similar mechanisms to induce IFN-I production [12].
TLR7, TLR8, and TLR9 also induce IFN-I expression, but only in plasmacytoid dendritic cells which are known to produce high levels of IFN after viral infection. These TLRs use the MYD88 adaptor which, in plasmacytoid dendritic cells, forms a complex with IRF7. This complex allows the phosphorylation and activation of IRF7 by interleukin 1 receptor associated kinase 1 (IRAK1) and triggers the expression of IFNs [17].
RLR pathway
Viral RNA binding on RLRs DDX58 and IFIH1 induces a conformational change of these receptors, which exposes their caspase activation and recruitment domains (CARD). These domains interact with the CARD of the mitochondrial antiviral signalling protein (MAVS, also called IPS-1). Subsequently, DDX58 and IFIH1 promote the formation of prion-like MAVS aggregates, which induce TRAF3 recruitment [18]. Several proteins of the RLR pathway are targeted by viruses. Influenza A virus (IAV) NS1 protein and respiratory syncytial virus NS1 protein bind DDX58 and MAVS, respectively, and block their signalling. IFIH1 is degraded following poliovirus infection, and encephalomyocarditis virus (EMCV) 3C protease can degrade DDX58 [10].
Activation of IRFs
TRAF3 recruits two kinases, TANK binding kinase 1 (TBK1) and inhibitor of nuclear factor kappa-B kinase subunit epsilon (IKKε), to phosphorylate and activate IRF3 and IRF7. Once phosphorylated, IRF3 and IRF7 form homodimers or heterodimers, translocate to the nucleus and promote IFN-I transcription [15, 19]. Viral proteins also target these factors. Ebola virus VP35 protein binds and blocks TBK1 and IKKε. The hepatitis C virus NS3/4A protease degrades IRF3 while viral homologues of IRFs, such as Kaposi’s sarcoma-associated herpesvirus vIRFs, bind host IRFs and inhibit IFN-I transcription [10].
IFNβ expression is regulated by four positive regulatory domains (PRD). NFκB and AP1 bind PRDII and PRDIV, respectively, and promote basal expression of IFNB1. After viral infection, IRF3 and IRF7 are activated and bind PRDI and PRDIII to induce IFNB1 overexpression [20]. IFNα genes have only PRDI- and PRDIII-like elements and their expression is therefore controlled exclusively by IRF3 and IRF7. IRF3 has more affinity for IFNB1 while IRF7 has more affinity for IFNα genes. IRF3 is constitutively abundant but inactive while IRF7 is an ISG present at low levels before infection and up-regulated by IFN-I signalling. Therefore, in the early phase after infection, IFN-I expression is induced by IRF3, resulting in predominant IFNβ production. IFNβ signalling induces IRF7 expression resulting in IFNα production in a later phase [8]. IRF1 and IRF5 can also induce IFN-I expression, however both are dispensable and their role remains unclear [8]. Furthermore, TLRs and RLRs also activate the NFκB pathway after infection through TICAM1, MYD88 and MAVS to induce the production of inflammatory cytokines [15].
Genetic susceptibility to viral infections
Several of the genes described above have been associated with susceptibilities to viral infections. These studies are summarized in Table 1 and Table 2 for human and mouse genes, respectively. Human studies split into case studies and association studies. Case studies aim to identify mutations, which strongly impact the severity of viral infection but are rare in the population. Association studies seek common genetic variants generally associated with a moderate impact. Genome-wide association studies require the analysis of large cohorts, which can rarely be assembled in infectious diseases. However, statistical power is increased by limiting the variants tested to a reduced set of candidate genes. This approach has led Zhang et al. to identify association between variants at 13 loci governing TLR3- and IRF7-dependent IFN-I immunity and the severity of COVID-19 by comparing 659 patients with life-threatening pneumonia and 534 patients with mild or no symptoms [21]. Likewise, Bigham et al. investigated 86 genes regulating immune function and identified association between three of them and the severity of West Nile virus (WNV) infection [22].
In mice, forward and reverse genetics are used to analyze resistance to viral infections [23]. Reverse genetics aims at characterizing the function of a given gene by altering its sequence. Many studies have reported modified susceptibility to viral infections in mice carrying loss-of-function mutations (gene knock-outs, KO) in IFN-I pathway. Forward genetics starts with a difference of susceptibility between two strains and aims at identifying the causal genetic variants. Differences may result from random chemical mutagenesis [24] or from natural variants between genetically diverse mouse strains such as the Collaborative Cross [6]. Interestingly, studies performed on the same virus can be compared to assess the specific or overlapping roles of the genes of the IFN-I cascade in the severity of a viral infection and in its complications.
TLR-TICAM1 pathway
As TLR3 is the primary TLR involved in IFN-I expression after virus recognition, the effects of its variants on the susceptibility to viral infections were extensively studied. In humans, association studies and case studies identified TLR3 variants linked to increased susceptibility to IAV [25,26,27,28], hepatitis B virus [29], herpes simplex virus 1 (HSV-1) [30,31,32], measles virus [33] and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [21]. Two SNPs associated with susceptibility to IAV are in intronic regions upstream exon 4 [25, 26]. Since this exon contains the signal induction transmembrane protein domain, these SNPs might alter TLR3 signalling. Other variants are in the luminal leucine-rich repeats of TLR3 [21, 27, 28, 30, 32, 33]. This region forms a solenoid critical for RNA binding [32] and virus recognition. Lastly, mutations were identified in the TIR domain of TLR3 [21, 30, 31]. In particular, a non-sense mutation was identified in a case of herpes simplex encephalitis (HSE), a complication of HSV-1 infection. This mutation removes the TIR domain, which is required for the recruitment of TICAM1 and downstream signalling [30].
In contrast, a common variant in TLR3 was associated with increased resistance to human immunodeficiency virus (HIV). The L412F allele, present in ~30% of Europeans and over-represented in a cohort of HIV-exposed seronegative individuals, leads to reduced viral replication and overexpression of inflammatory cytokines in vitro [34], likely by increasing TLR3 signalling. However, the same variant showed positive association with subacute sclerosing panencephalitis, a severe complication of measles virus infection [33]. The increased inflammatory response due to this mutation may be advantageous in the case of HIV infection, but deleterious in the case of measles virus infection. Variants in the TLR adaptor TICAM1 can also alter susceptibility to viruses. In humans, four mutations were identified in patients suffering from HSE and three in patients with life-threatening COVID-19, all of them leading to decreased IFN-I expression [21, 35, 36].
The TLR-TICAM1 pathway has been also extensively studied in mouse viral infections. Compared to wild-type (WT) mice, Tlr3-deficient mice showed a decreased survival rate with higher viral loads in coxsackievirus B3 [37] and EMCV [38] infections and, while they showed an increased serum viral load but unchanged mortality after murine cytomegalovirus (MCMV) infection [39]. In contrast, Tlr3-deficient mice displayed a decreased mortality following IAV infection [40]. Mice carrying a frameshift-induced deletion in the Ticam1 gene showed enhanced susceptibility to MCMV with increased viral load in the spleen and higher mortality [41]. Another study found that Ticam1-deficient mice were more susceptible to CVB3. Interestingly, these mice presented a decreased IFN-I expression 72 h post-infection, but an increased expression 7 days after infection [42], which may result from an uncontrolled inflammatory response. Tlr3-deficient mice also developed cardiac anomalies, a complication of CVB3 infection, with large myocarditic lesions and increased heart viral load [37]. Similarly, Ticam1-deficient mice presented left ventricular dysfunction and severe myocardial damage including cardiac fibrosis. These mice also showed increased heart viral load [42]. The overlapping phenotypes observed in these two studies are consistent with the direct interactions between Tlr3 and Ticam1 in the IFN-I induction cascade.
Tlr3-deficient mice were also less susceptible to vaccinia virus (VV) infection than WT mice with higher viral load, while Ticam1- and Tlr4-deficient mice were more susceptible [43, 44]. It was hypothesized that abrogating Tlr3 signaling decreases the inflammatory response and thus the complications resulting from VV infection. In contrast, since Tlr4 signaling activates IRFs and NFκB, Tlr4 and Ticam1 KOs block both pathways and lead to increased susceptibility to VV infection [43, 44].
The outcome of WNV infection in Tlr3-deficient mice was investigated in two studies, which used the same mouse strain and two closely related virus strains with contrasted results. Wang et al. reported that Tlr3-deficient mice presented a decreased mortality after infection with WNV isolate 2741, but an increased viral load. Moreover, these mice showed decreased neuronal inflammation and blood-brain barrier permeability, suggesting that Tlr3 is involved in the virus brain entry [45]. Daffis et al. who used the WNV strain 3000.0259 reported that Tlr3-deficient mice also presented a higher brain viral load but with susceptibility to WNV infection and mortality than WT mice. Unlike the previous study, blood-brain barrier permeability and neuroinflammation were not affected, compared with WT mice [46]. These contrasted results were attributed to the infection route, the viral dose and the cells used to produce the virus which differed between the two studies [46]. Interestingly, Tlr3-deficient mice produced normal amount of IFN-I in the first study, while they were decreased in the second study, leading to the hypothesis that IFN-I expression could also have a detrimental effect in WNV infection [45]. These results illustrate the dual role of Tlr3 signalling which may lead to an excessive inflammatory response, while decreased inflammation in Tlr3 KO mice may reduce the risk of severe complication.
TLR-MYD88 pathway
In mice, deficiency in Tlr2, Tlr4, Tlr7 or Tlr9 was associated with increased or decreased susceptibility to viral infections. However, since these receptors signal through the MYD88 adaptor which also activates the NFκB pathway, their role in the susceptibility to viruses may not be solely associated with the IFN-I pathway.
Tlr7-deficient mice were more susceptible to WNV infection [47]. However, they presented an increased IFN-I expression which could result from the signaling through other receptors such as Tlr3 and RLRs. Tlr9 and Myd88-deficient mice were more susceptible to MCMV with decreased IFN-I production [39]. Myd88 KO mice had a reduced number of splenic plasmacytoid dendritic cells which could explain reduced levels of IFN-I. By contrast, Tlr2 KO mice were less susceptible to HSV-1 with reduced mortality compared to WT mice. They also showed decreased NFκB-induced cytokine production, which may explain a milder inflammatory state and the absence of severe complications [48]. IFN-I expression was not investigated although it could contribute to the pathology.
Myd88 deficiency in mice also resulted in increased susceptibility to chikungunya virus (CHIKV) and severe acute respiratory syndrome coronavirus (SARS-CoV). Indeed, Myd88 KO mice presented higher viral loads following CHIKV infection [49], and higher mortality and increased viral load following SARS-CoV infection [50]. Two studies with WNV led to similar results [47, 51]. Interestingly, in one study, Myd88 KO mice had higher levels of IFN-I after infection than WT mice due to an increased expression in bone marrow-derived macrophages. This might result from high viral replication in these cells and from the signaling of other pathways, such as RLR- or Tlr3-dependent pathways [51]. The role of Ticam2 in the susceptibility to SARS-CoV was suspected in an association study using the Collaborative Cross and was confirmed with a Ticam2-deficient strain which showed higher lung viral loads than WT mice [52].
RLR pathway
In humans, two variants were identified in DDX58 in a patient who suffered from severe IAV infection. The R71H variant is in the CARD protein domain, while the P885 variant is in the regulatory domain involved in viral RNA recognition. These variants lead to impaired IFN-I expression following IAV infection when expressed in DDX58 deficient human embryonic kidney cells 293, but not in the patient’s peripheral blood mononuclear cells where other pathways, such as TLR7-dependant signalling might ensure a correct expression [53]. Variants were identified in IFIH1 in patients suffering from bronchiolitis following rhinovirus or respiratory syncytial virus infection and led to decreased expression of IFNβ [54]. Moreover, the K365E mutation was identified in a 5-year-old child suffering from numerous recurrent respiratory virus infections. This mutation prevents IFIH1 from interacting with viral RNA, thus inhibiting IFN-I induction [55]. The H843A mutation in IFIH1 was also associated with susceptibility to HCV by comparing patients with spontaneously resolved hepatitis or chronic hepatitis [56]. In two studies, Ddx58-deficient mice showed similar mortality after IAV infection compared with WT individuals [57, 58]. Notably, one study showed that Ddx58 deficiency also led to defects in adaptive immunity affecting antigen presentation by dendritic cells and activation of T cell responses [57]. Ifih1-deficient mice were more susceptible to mouse hepatitis virus. Interestingly, they showed decreased expression of IFN-I but normal induction of ISGs [59]. Moreover, they were found to be more susceptible to human metapneumovirus [60], to hepatitis B virus [61], to murine norovirus 1 [62], and more prone to develop demyelinating disease following Theiler’s murine encephalomyelitis virus infection [63].
In mice, RLRs and MAVS have often been studied together, which has unraveled their specificity. Using Ddx58- and Ifih1-deficient mouse embryonic fibroblasts (MEFs) infected with several viruses, Kato et al. found that these two receptors recognize different viruses. Moreover, they showed that Ddx58- and Ifih1-deficient mice were more susceptible to Japanese encephalitis virus than WT mice, and that Ifih1- but not Ddx58-deficient mice were more susceptible to EMCV [64]. Susceptibility of Ifih1-deficient mice to EMCV infection was also reported in another study [65]. Furthermore, Ddx58-deficient mice showed increased serum viral load following CHIKV infection, which was not the case for Ifih1-deficient mice [49]. These results show that Ddx58 and Ifih1 have complementary roles in the recognition of viral RNA, consistently with their known differences in molecular pattern recognition.
By contrast, both Ddx58- and Ifih1-deficient mice showed increased susceptibility to WNV. Double-deficient mice were even more susceptible and invariably died within 8 days after infection, showing that both receptors are involved in the recognition of WNV. The phenotype of double-deficient mice was very similar to that of Mavs-deficient mice through which both RLRs signal [66]. Indeed, Mavs deficiency resulted in increased susceptibility to WNV with higher mortality and viral load, and deficient activation of IFNβ [67]. Mavs and Ifih1 deficiencies resulted also in increased mortality following CVB3 infection and decreased expression of IFN-I although viral titers were identical to WT mice [68]. Mavs deficiency also resulted in increased susceptibility to EMCV [69], to vesicular stomatitis virus [69, 70] and to dengue virus (DENV) [71], and in increased serum viral load following CHIKV infection [49].
Mice deficient for the auxiliary RLR Dhx58 gene also showed increased susceptibility to EMCV [13] and to WNV [14]. WNV-infected, Dhx58-deficient mice showed increased mortality but similar kinetics of IFNβ production and tissue viral loads compared with WT mice. In the brain they displayed increased neuronal damage, elevated viral load in a late phase of infection, low neuroinflammation and decreased recruitment of CD8 + T cells [14]. Dhx58 is therefore required for protection against WNV infection. Furthermore, mice overexpressing Dhx58 were more resistant to IAV infection [72]. In vitro, Dhx58-deficient cells exposed to several RNA viruses produced less IFN-I than WT cells suggesting that Dhx58 is required for Ddx58- and Ifih1-mediated antiviral responses [13].
Activation of IRFs and IFNs
Sequencing of candidate genes in HSE patients identified a heterozygous missense mutation in TRAF3 associated with decreased IFN-I expression [73] and two missense mutations in TBK1 affecting the kinase domain and thus preventing the phosphorylation of target proteins [74]. These mutations resulted in reduced IFN-I expression in cells stimulated with synthetic RNA. Two dominant mutations in TBK1 were identified in patients with severe COVID-19 and led to decreased IFN-I expression in HEK293T cells transfected with these mutant forms of TBK1 [21].
In mice, no genetic variants in Traf3 or in Tbk1 have been associated with altered susceptibility to viral infections. However, mice deficient for the Ikbke gene (encoding IKKε, a kinase involved in IRFs activation) showed extreme susceptibility to IAV despite normal expression of IFN-I. Mechanistic studies revealed that IKKε indirectly controls the expression of a subset of ISGs [75].
Mutations in IRFs have been repeatedly associated with susceptibility to viral infections in humans and in mice. In humans, two missense mutations were found in IRF3 in patients suffering from HSE [35, 76]. These mutations are located in the IRF association domain and might therefore prevent IRF3 dimerization [77]. Two autosomal dominant mutations in IRF3 were identified in COVID-19 patients with pneumonia [21] and a non-coding variant was associated with susceptibility to WNV by comparing asymptomatic and symptomatic infected individuals [22]. A compound heterozygosity was found in IRF7 in a patient suffering from life-threatening infection following IAV infection [78] and seven mutations in IRF7 were found in COVID-19 patients leading to decreased IFN-I induction [21].
In mice, Irf3 and Irf7 deficiencies have been studied in isolation or in combination. Both single deficiencies increased mortality following IAV infection and susceptibility was further enhanced in double deficient mice [79, 80]. Viral load in lungs was not significantly altered in Irf7-deficient mice but was increased in Irf3-deficient mice and even more in double-deficient mice, suggesting that Irf7 also contributes to controlling viral replication. On day 2 after infection, IFNα expression was reduced in Irf3- but not in Irf7-deficient mice while IFNβ expression was reduced in Irf7 but not in Irf3-deficient mice [79]. This result is consistent with the distinct affinities of the two IRFs for the IFN-I genes. Mice deficient for Irf3 and Irf7 were also susceptible to WNV with increased mortality rate and viral load, decreased expression of IFN-I and increased viral load in the brain [81, 82].
However, the consequences of Irf3 and Irf7 deficiencies are variable between viruses. Irf7- but not Irf3-deficient mice were susceptible to HSV-1 and they were more susceptible to EMCV than Irf3-deficient mice [83]. In the case of CHIKV infection, Irf3-Irf7 double KO mice were highly susceptible with increased viremia and mortality, while Irf3- and Irf7-deficient mice survived and had normal viremia [84]. Following DENV infection, Irf3-Irf7 double KO mice and Irf7-deficient mice showed increased viral load and decreased IFN-I expression but survived the infection [85]. In the case of Zika virus infection, Irf3-Irf5-Irf7 triple KO mice died with neurological disease signs, while Irf3-deficient mice survived [86]. Interestingly, Irf3-Irf5-Irf7 triple KO mice survived to DENV infection through robust induction of type II IFNs, but showed increased viremia. This resistance to DENV was abolished when Irf1 was also inactivated, which led to the identification of a protective Irf1-dependent pathway [87].
Lastly, variants in IFN-I genes themselves were associated with susceptibility to viral diseases in mice. The role of IFN-I in viral infections has been extensively investigated using mice deficient for their receptor. Ifnar1-deficient mice showed increased susceptibility to a number of viruses including CHIKV [49], Zika virus [86], DENV [71] and Ebola virus [88]. Transient blockade of IFNα and IFNβ with monoclonal antibodies resulted in increased mortality after WNV infection [89]. WNV susceptibility was also studied in Ifnb1−/− mice and led to similar results [90]. IFNβ-deficient mice were also found more susceptible to VV than WT mice [91]. These two studies led to opposite results regarding IFNα expression. The absence of IFNβ is expected to abrogate Irf7 induction and thus to decrease IFNα expression. This was indeed observed after VV infection. However, after WNV infection, IFNα was upregulated, which was hypothesized to result from the high viral load [90]. IFNβ-deficient mice also showed increased susceptibility to IAV [92] and CVB3 [93], and increased spleen viral load following Friend virus infection [94].
Discussion
Type I IFNs are critical components of the immediate response against invading viruses. Indeed, their induction allows the expression of many ISGs which can control viral infection. The pathway leading to IFN-I production is complex as many genes are involved, and viral proteins target this pathway at multiple levels. Moreover, some of these genes, such as IRF7, are also ISGs, which further complicates the kinetics of IFN-I activation. Despite the vast number of studies carried out on the induction of IFN-I, not all mechanisms are yet fully understood.
In accordance with the functions of IFN-I, mutations in most genes of the induction pathway have been associated with increased susceptibility to viral infections in human and mice. In humans, whole exome or candidate gene sequencing has identified coding and non-coding variants, primarily in patients with severe forms of infections. It is likely that other variants are present in the human population but the power to detect them in association studies depends on their frequency, on their impact on host response to infections and on cohort size. In mice, most studies have used reverse genetics approaches and have investigated the consequences of complete loss-of-function mutations in infected mice which, in most cases, led to higher susceptibility, with mortality and elevated viral load in tissues. While all mutants reported here were constitutively deficient, tissue-specific conditional alleles allow investigating the pathway in specific cell lineages. For example, myeloid-conditional Tbk1-deficient mice showed increased survival to IAV infection with reduced inflammation in the respiratory tract, demonstrating the role of myeloid cells in disease pathophysiology [95].
Notably, a few genes of the pathways were not tested by reverse genetics. Tlr2 and Tlr4 have been investigated mostly for their role in bacterial infections [11]. For other genes, like Tbk1 and Traf3, deficiency was only studied in vitro on MEFs or macrophages since homozygous mice die either in utero (Tkb1) or a few days after birth (Traf3) [96]. Interestingly, Marchlik et al. produced a Tbk1 mutation which resulted in a catalytically inactive protein and they could obtain homozygous deficient mice with complete ablation of IFNβ production [97]. This difference in survival of Tbk1-deficient mice is likely due to the 129S5 genetic background on which this allele was created, compared with the C57BL/6 background used for most KO alleles. This case highlights the importance of mouse genetic background when evaluating the phenotype resulting from gene inactivation [98].
The formal description of the IFN-I activation cascade incompletely reflects the complexity of the mechanisms from viral components recognition to IFN-I-induced effectors. As exemplified by Irf3 and Irf7, the effect of a host gene variant may be different between viruses. Moreover, while deficiency of most pathway genes resulted in increased susceptibility to viral infections due to impaired IFNβ production, it could also be associated with reduced susceptibility as in the case of Tlr3. This observation underlines the complexity of immune mechanisms, and the importance of balanced and well-controlled IFN response. While rapid activation of ISGs is critical to the control of viral replication, excessive or persistent IFN−Ι production can be detrimental by triggering inflammatory processes responsible for tissue damage and organ failure. Notably, dysregulation of immune responses with delayed expression of IFN-I and robust cytokine response could be at the origin of the clinical manifestations observed in severe SARS-CoV [99] and SARS-CoV-2 infections [100].
Investigating the role of every gene of the IFN-I induction cascade by gene inactivation has contributed to dissecting the mechanisms of the pathway. However, a non-functional step may result from defective interactions between functional but incompatible partner proteins. Such interactions could occur for example if the two partners were inherited from genetically distant parents. In mice, investigating strains produced by crosses between founders of different subspecific origins, like the Collaborative Cross, may identify such situations and provide new variants for functional analysis [6]. With the growing evidence that microbiota can also modify the IFN-I response and therefore the susceptibility to infectious diseases [101], it is clear that we are still far from understanding the subtle regulations of an essential pathway.
References
Fensterl V, Sen GC. Interferons and viral infections. Biofactors. 2009;35:14–20.
Beachboard DC, Horner SM. Innate immune evasion strategies of DNA and RNA viruses. Curr Opin Microbiol. 2016;32:113–9.
Lee AJ, Ashkar AA. The Dual Nature of Type I and Type II Interferons. Front Immunol. 2018;9:2061.
Zhou JH, Wang YN, Chang QY, Ma P, Hu Y, Cao X. Type III Interferons in Viral Infection and Antiviral Immunity. Cell Physiol Biochem. 2018;51:173–85.
Kenney AD, Dowdle JA, Bozzacco L, McMichael TM, St Gelais C, Panfil AR, et al. Human Genetic Determinants of Viral Diseases. Annu Rev Genet. 2017;51:241–63.
Noll KE, Ferris MT, Heise MT. The collaborative cross: a systems genetics resource for studying host-pathogen interactions. Cell Host Microbe. 2019;25:484–98.
Hardy MP, Owczarek CM, Jermiin LS, Ejdeback M, Hertzog PJ. Characterization of the type I interferon locus and identification of novel genes. Genomics. 2004;84:331–45.
Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–60.
Li SF, Gong MJ, Zhao FR, Shao JJ, Xie YL, Zhang YG, et al. Type I interferons: distinct biological activities and current applications for viral infection. Cell Physiol Biochem. 2018;51:2377–96.
Nan Y, Nan G, Zhang YJ. Interferon induction by RNA viruses and antagonism by viral pathogens. Viruses. 2014;6:4999–5027.
Lester SN, Li K. Toll-like receptors in antiviral innate immunity. J Mol Biol. 2014;426:1246–64.
Stack J, Doyle SL, Connolly DJ, Reinert LS, O’Keeffe KM, McLoughlin RM, et al. TRAM is required for TLR2 endosomal signaling to type I IFN induction. J Immunol. 2014;193:6090–102.
Satoh T, Kato H, Kumagai Y, Yoneyama M, Sato S, Matsushita K, et al. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc Natl Acad Sci USA. 2010;107:1512–7.
Suthar MS, Ramos HJ, Brassil MM, Netland J, Chappell CP, Blahnik G, et al. The RIG-I-like receptor LGP2 controls CD8(+) T cell survival and fitness. Immunity. 2012;37:235–48.
Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461.
Funami K, Matsumoto M, Oshiumi H, Inagaki F, Seya T. Functional interfaces between TICAM-2/TRAM and TICAM-1/TRIF in TLR4 signaling. Biochem Soc Trans. 2017;45:929–35.
Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131–7.
Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–61.
Ning S, Pagano JS, Barber GN. IRF7: activation, regulation, modification and function. Genes Immun. 2011;12:399–414.
Levy DE, Marie IJ, Durbin JE. Induction and function of type I and III interferon in response to viral infection. Curr Opin Virol. 2011;1:476–86.
Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370:eabd4570.
Bigham AW, Buckingham KJ, Husain S, Emond MJ, Bofferding KM, Gildersleeve H, et al. Host genetic risk factors for West Nile virus infection and disease progression. PLoS ONE. 2011;6:e24745.
Beutler B, Eidenschenk C, Crozat K, Imler JL, Takeuchi O, Hoffmann JA, et al. Genetic analysis of resistance to viral infection. Nat Rev Immunol. 2007;7:753–66.
Crozat K, Georgel P, Rutschmann S, Mann N, Du X, Hoebe K, et al. Analysis of the MCMV resistome by ENU mutagenesis. Mamm Genome. 2006;17:398–406.
Esposito S, Molteni CG, Giliani S, Mazza C, Scala A, Tagliaferri L, et al. Toll-like receptor 3 gene polymorphisms and severity of pandemic A/H1N1/2009 influenza in otherwise healthy children. Virol J. 2012;9:270.
Lee N, Cao B, Ke C, Lu H, Hu Y, Tam CHT, et al. IFITM3, TLR3, and CD55 Gene SNPs and Cumulative Genetic Risks for Severe Outcomes in Chinese Patients With H7N9/H1N1pdm09 Influenza. J Infect Dis. 2017;216:97–104.
Hidaka F, Matsuo S, Muta T, Takeshige K, Mizukami T, Nunoi H. A missense mutation of the Toll-like receptor 3 gene in a patient with influenza-associated encephalopathy. Clin Immunol. 2006;119:188–94.
Lim HK, Huang SXL, Chen J, Kerner G, Gilliaux O, Bastard P, et al. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J Exp Med. 2019;216:2038–56.
Al-Qahtani A, Al-Ahdal M, Abdo A, Sanai F, Al-Anazi M, Khalaf N, et al. Toll-like receptor 3 polymorphism and its association with hepatitis B virus infection in Saudi Arabian patients. J Med Virol. 2012;84:1353–9.
Guo Y, Audry M, Ciancanelli M, Alsina L, Azevedo J, Herman M, et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J Exp Med. 2011;208:2083–98.
Lim HK, Seppanen M, Hautala T, Ciancanelli MJ, Itan Y, Lafaille FG, et al. TLR3 deficiency in herpes simplex encephalitis: high allelic heterogeneity and recurrence risk. Neurology. 2014;83:1888–97.
Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–7.
Ishizaki Y, Takemoto M, Kira R, Kusuhara K, Torisu H, Sakai Y, et al. Association of toll-like receptor 3 gene polymorphism with subacute sclerosing panencephalitis. J Neurovirol. 2008;14:486–91.
Sironi M, Biasin M, Cagliani R, Forni D, De Luca M, Saulle I, et al. A common polymorphism in TLR3 confers natural resistance to HIV-1 infection. J Immunol. 2012;188:818–23.
Mork N, Kofod-Olsen E, Sorensen KB, Bach E, Orntoft TF, Ostergaard L, et al. Mutations in the TLR3 signaling pathway and beyond in adult patients with herpes simplex encephalitis. Genes Immun. 2015;16:552–66.
Sancho-Shimizu V, Perez de Diego R, Lorenzo L, Halwani R, Alangari A, Israelsson E, et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J Clin Investig. 2011;121:4889–902.
Negishi H, Osawa T, Ogami K, Ouyang X, Sakaguchi S, Koshiba R, et al. A critical link between Toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc Natl Acad Sci USA. 2008;105:20446–51.
Hardarson HS, Baker JS, Yang Z, Purevjav E, Huang CH, Alexopoulou L, et al. Toll-like receptor 3 is an essential component of the innate stress response in virus-induced cardiac injury. Am J Physiol Heart Circ Physiol. 2007;292:H251–8.
Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci USA. 2004;101:3516–21.
Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, et al. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2006;2:e53.
Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–8.
Riad A, Westermann D, Zietsch C, Savvatis K, Becher PM, Bereswill S, et al. TRIF is a critical survival factor in viral cardiomyopathy. J Immunol. 2011;186:2561–70.
Hutchens M, Luker KE, Sottile P, Sonstein J, Lukacs NW, Nunez G, et al. TLR3 increases disease morbidity and mortality from vaccinia infection. J Immunol. 2008;180:483–91.
Hutchens MA, Luker KE, Sonstein J, Nunez G, Curtis JL, Luker GD. Protective effect of Toll-like receptor 4 in pulmonary vaccinia infection. PLoS Pathog. 2008;4:e1000153.
Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366–73.
Daffis S, Samuel MA, Suthar MS, Gale M Jr., Diamond MS. Toll-like receptor 3 has a protective role against West Nile virus infection. J Virol. 2008;82:10349–58.
Town T, Bai F, Wang T, Kaplan AT, Qian F, Montgomery RR, et al. Toll-like receptor 7 mitigates lethal West Nile encephalitis via interleukin 23-dependent immune cell infiltration and homing. Immunity. 2009;30:242–53.
Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, et al. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci USA. 2004;101:1315–20.
Schilte C, Couderc T, Chretien F, Sourisseau M, Gangneux N, Guivel-Benhassine F, et al. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J Exp Med. 2010;207:429–42.
Sheahan T, Morrison TE, Funkhouser W, Uematsu S, Akira S, Baric RS, et al. MyD88 is required for protection from lethal infection with a mouse-adapted SARS-CoV. PLoS Pathog. 2008;4:e1000240.
Szretter KJ, Daffis S, Patel J, Suthar MS, Klein RS, Gale M Jr., et al. The innate immune adaptor molecule MyD88 restricts West Nile virus replication and spread in neurons of the central nervous system. J Virol. 2010;84:12125–38.
Gralinski LE, Menachery VD, Morgan AP, Totura AL, Beall A, Kocher J, et al. Allelic Variation in the Toll-Like Receptor Adaptor Protein Ticam2 Contributes to SARS-Coronavirus Pathogenesis in Mice. G3 (Bethesda). 2017;7:1653–63.
Jorgensen SE, Christiansen M, Ryo LB, Gad HH, Gjedsted J, Staeheli P, et al. Defective RNA sensing by RIG-I in severe influenza virus infection. Clin Exp Immunol. 2018;192:366–76.
Asgari S, Schlapbach LJ, Anchisi S, Hammer C, Bartha I, Junier T, et al. Severe viral respiratory infections in children with IFIH1 loss-of-function mutations. Proc Natl Acad Sci USA. 2017;114:8342–7.
Lamborn IT, Jing H, Zhang Y, Drutman SB, Abbott JK, Munir S, et al. Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J Exp Med. 2017;214:1949–72.
Hoffmann FS, Schmidt A, Dittmann Chevillotte M, Wisskirchen C, Hellmuth J, Willms S, et al. Polymorphisms in melanoma differentiation-associated gene 5 link protein function to clearance of hepatitis C virus. Hepatology. 2015;61:460–70.
Kandasamy M, Suryawanshi A, Tundup S, Perez JT, Schmolke M, Manicassamy S, et al. RIG-I Signaling Is Critical for Efficient Polyfunctional T Cell Responses during Influenza Virus Infection. PLoS Pathog. 2016;12:e1005754.
Wu W, Wang X, Zhang W, Tian L, Booth JL, Duggan ES, et al. RIG-I Signaling via MAVS Is Dispensable for Survival in Lethal Influenza Infection In Vivo. Mediators Inflamm. 2018;2018:6808934.
Zalinger ZB, Elliott R, Rose KM, Weiss SR. MDA5 Is Critical to Host Defense during Infection with Murine Coronavirus. J Virol. 2015;89:12330–40.
Banos-Lara Mdel R, Ghosh A, Guerrero-Plata A. Critical role of MDA5 in the interferon response induced by human metapneumovirus infection in dendritic cells and in vivo. J Virol. 2013;87:1242–51.
Lu HL, Liao F. Melanoma differentiation-associated gene 5 senses hepatitis B virus and activates innate immune signaling to suppress virus replication. J Immunol. 2013;191:3264–76.
McCartney SA, Thackray LB, Gitlin L, Gilfillan S, Virgin HW, Colonna M. MDA-5 recognition of a murine norovirus. PLoS Pathog. 2008;4:e1000108.
Jin YH, Kim SJ, So EY, Meng L, Colonna M, Kim BS. Melanoma differentiation-associated gene 5 is critical for protection against Theiler’s virus-induced demyelinating disease. J Virol. 2012;86:1531–43.
Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–5.
Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, et al. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA. 2006;103:8459–64.
Errett JS, Suthar MS, McMillan A, Diamond MS, Gale M Jr. The essential, nonredundant roles of RIG-I and MDA5 in detecting and controlling West Nile virus infection. J Virol. 2013;87:11416–25.
Suthar MS, Ma DY, Thomas S, Lund JM, Zhang N, Daffis S, et al. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog. 2010;6:e1000757.
Wang JP, Cerny A, Asher DR, Kurt-Jones EA, Bronson RT, Finberg RW. MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J Virol. 2010;84:254–60.
Kumar H, Kawai T, Kato H, Sato S, Takahashi K, Coban C, et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J Exp Med. 2006;203:1795–803.
Sun Q, Sun L, Liu HH, Chen X, Seth RB, Forman J, et al. The specific and essential role of MAVS in antiviral innate immune responses. Immunity. 2006;24:633–42.
Perry ST, Prestwood TR, Lada SM, Benedict CA, Shresta S. Cardif-mediated signaling controls the initial innate response to dengue virus in vivo. J Virol. 2009;83:8276–81.
Si-Tahar M, Blanc F, Furio L, Chopy D, Balloy V, Lafon M, et al. Protective role of LGP2 in influenza virus pathogenesis. J Infect Dis. 2014;210:214–23.
Perez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity. 2010;33:400–11.
Herman M, Ciancanelli M, Ou YH, Lorenzo L, Klaudel-Dreszler M, Pauwels E, et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J Exp Med. 2012;209:1567–82.
Tenoever BR, Ng SL, Chua MA, McWhirter SM, Garcia-Sastre A, Maniatis T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science. 2007;315:1274–8.
Andersen LL, Mork N, Reinert LS, Kofod-Olsen E, Narita R, Jorgensen SE, et al. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J Exp Med. 2015;212:1371–9.
Lin R, Mamane Y, Hiscott J. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol Cell Biol. 1999;19:2465–74.
Ciancanelli MJ, Huang SX, Luthra P, Garner H, Itan Y, Volpi S, et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science. 2015;348:448–53.
Hatesuer B, Hoang HT, Riese P, Trittel S, Gerhauser I, Elbahesh H, et al. Deletion of Irf3 and Irf7 Genes in Mice Results in Altered Interferon Pathway Activation and Granulocyte-Dominated Inflammatory Responses to Influenza A Infection. J Innate Immun. 2017;9:145–61.
Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–48.
Daffis S, Samuel MA, Keller BC, Gale M Jr., Diamond MS. Cell-specific IRF-3 responses protect against West Nile virus infection by interferon-dependent and -independent mechanisms. PLoS Pathog. 2007;3:e106.
Daffis S, Samuel MA, Suthar MS, Keller BC, Gale M Jr., Diamond MS. Interferon regulatory factor IRF-7 induces the antiviral alpha interferon response and protects against lethal West Nile virus infection. J Virol. 2008;82:8465–75.
Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–7.
Rudd PA, Wilson J, Gardner J, Larcher T, Babarit C, Le TT, et al. Interferon response factors 3 and 7 protect against Chikungunya virus hemorrhagic fever and shock. J Virol. 2012;86:9888–98.
Chen HW, King K, Tu J, Sanchez M, Luster AD, Shresta S. The roles of IRF-3 and IRF-7 in innate antiviral immunity against dengue virus. J Immunol. 2013;191:4194–201.
Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, et al. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe. 2016;19:720–30.
Carlin AF, Plummer EM, Vizcarra EA, Sheets N, Joo Y, Tang W, et al. An IRF-3-, IRF-5-, and IRF-7-Independent Pathway of Dengue Viral Resistance Utilizes IRF-1 to Stimulate Type I and II Interferon Responses. Cell Rep. 2017;21:1600–12.
Brannan JM, Froude JW, Prugar LI, Bakken RR, Zak SE, Daye SP, et al. Interferon alpha/beta Receptor-Deficient Mice as a Model for Ebola Virus Disease. J Infect Dis. 2015;212:S282–94.
Sheehan KC, Lazear HM, Diamond MS, Schreiber RD. Selective Blockade of Interferon-alpha and -beta Reveals Their Non-Redundant Functions in a Mouse Model of West Nile Virus Infection. PLoS ONE. 2015;10:e0128636.
Lazear HM, Pinto AK, Vogt MR, Gale M Jr., Diamond MS. Beta interferon controls West Nile virus infection and pathogenesis in mice. J Virol. 2011;85:7186–94.
Deonarain R, Alcami A, Alexiou M, Dallman MJ, Gewert DR, Porter AC. Impaired antiviral response and alpha/beta interferon induction in mice lacking beta interferon. J Virol. 2000;74:3404–9.
Koerner I, Kochs G, Kalinke U, Weiss S, Staeheli P. Protective role of beta interferon in host defense against influenza A virus. J Virol. 2007;81:2025–30.
Deonarain R, Cerullo D, Fuse K, Liu PP, Fish EN. Protective role for interferon-beta in coxsackievirus B3 infection. Circulation. 2004;110:3540–3.
Gerlach N, Schimmer S, Weiss S, Kalinke U, Dittmer U. Effects of type I interferons on Friend retrovirus infection. J Virol. 2006;80:3438–44.
Hagan RS, Torres-Castillo J, Doerschuk CM. Myeloid TBK1 Signaling Contributes to the Immune Response to Influenza. Am J Respir Cell Mol Biol. 2019;60:335–45.
Lalani AI, Moore CR, Luo C, Kreider BZ, Liu Y, Morse HC 3rd, et al. Myeloid cell TRAF3 regulates immune responses and inhibits inflammation and tumor development in mice. J Immunol. 2015;194:334–48.
Marchlik E, Thakker P, Carlson T, Jiang Z, Ryan M, Marusic S, et al. Mice lacking Tbk1 activity exhibit immune cell infiltrates in multiple tissues and increased susceptibility to LPS-induced lethality. J Leukoc Biol. 2010;88:1171–80.
Manet C, Simon-Loriere E, Jouvion G, Hardy D, Prot M, Conquet L et al. Genetic Diversity of Collaborative Cross Mice Controls Viral Replication, Clinical Severity, and Brain Pathology Induced by Zika Virus Infection, Independently of Oas1b. J Virol. 2020;94:e01034–19.
Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe. 2016;19:181–93.
Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020;181:1036–45. e9.
Steed AL, Christophi GP, Kaiko GE, Sun L, Goodwin VM, Jain U, et al. The microbial metabolite desaminotyrosine protects from influenza through type I interferon. Science. 2017;357:498–502.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bourdon, M., Manet, C. & Montagutelli, X. Host genetic susceptibility to viral infections: the role of type I interferon induction. Genes Immun 21, 365–379 (2020). https://doi.org/10.1038/s41435-020-00116-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41435-020-00116-2
This article is cited by
-
Inborn Error of STAT2-Dependent IFN-I Immunity in a Patient Presented with Hemophagocytic Lymphohistiocytosis and Multisystem Inflammatory Syndrome in Children
Journal of Clinical Immunology (2023)
-
A systematic review and meta-analysis of host genetic factors associated with influenza severity
BMC Genomics (2021)
