
Abstract
Gene therapy for auditory diseases is gradually maturing. Recent progress in gene therapy treatments for genetic and acquired hearing loss has demonstrated the feasibility in animal models. However, a number of hurdles, such as lack of safe viral vector with high efficiency and specificity, robust deafness large animal models, translating animal studies to clinic etc., still remain to be solved. It is necessary to overcome these challenges in order to effectively recover auditory function in human patients. Here, we review the progress made in our group, especially our efforts to make more effective and cell type-specific viral vectors for targeting cochlea cells.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others
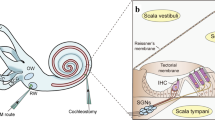

Change history
08 July 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41434-021-00274-6
10 August 2020
A Correction to this paper has been published: https://doi.org/10.1038/s41434-020-00186-x
References
Dai P, Huang LH, Wang GJ, Gao X, Qu CY, Chen XW, et al. Concurrent hearing and genetic screening of 180,469 neonates with follow-up in Beijing, China. Am J Hum Genet. 2019;105:803–12.
Akil O, Seal RP, Burke K, Wang C, Alemi A, During M, et al. Restoration of hearing in the VGLUT3 knockout mouse using virally mediated gene therapy. Neuron. 2012;75:283–93.
LeMasurier M, Gillespie PG. Hair-cell mechanotransduction and cochlear amplification. Neuron. 2005;48:403–15.
Ahmed H, Shubina-Oleinik O, Holt JR. Emerging gene therapies for genetic hearing loss. J Assoc Res Otolaryngol. 2017;18:649–70.
Géléoc GSG, Holt JR. Sound strategies for hearing restoration. Science. 2014;344:1241062.
Zhang W, Kim SM, Wang W, Cai C, Feng Y, Kong W, et al. Cochlear gene therapy for sensorineural hearing loss: current status and major remaining hurdles for translational success. Front Mol Neurosci. 2018;11:221.
Pan B, Akyuz N, Liu X-P, Asai Y, Nist-Lund C, Kurima K, et al. TMC1 forms the pore of mechanosensory transduction channels in vertebrate inner ear hair cells. Neuron. 2018;99:736–53.e6.
Roux I, Safieddine S, Nouvian R, Grati M, Simmler MC, Bahloul A, et al. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006;127:277–89.
Takago H, Oshima-Takago T, Moser T. Disruption of otoferlin alters the mode of exocytosis at the mouse inner hair cell ribbon synapse. Front Mol Neurosci. 2019;11:492–492.
Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJ, Steel KP, et al. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat Genet. 1997;16:188–90.
Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ, et al. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet. 2001;69:25–34.
Vahava O, Morell R, Lynch ED, Weiss S, Kagan ME, Ahituv N, et al. Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science. 1998;279:1950–4.
Xiang M, Maklad A, Pirvola U, Fritzsch B. Brn3c null mutant mice show long-term, incomplete retention of some afferent inner ear innervation. BMC Neurosci. 2003;4:2.
Hu J, Li B, Apisa L, Yu H, Entenman S, Xu M, et al. ER stress inhibitor attenuates hearing loss and hair cell death in Cdh23(erl/ erl) mutant mice. Cell Death Dis. 2016;7:e2485–e2485.
Zhang YP, Tang WX, Ahmad S, Sipp JA, Chen P, Lin X. Gap junction-mediated intercellular biochemical coupling in cochlear supporting cells is required for normal cochlear functions. P Natl Acad Sci USA. 2005;102:15201–6.
Park H-J, Houn Hahn S, Chun Y-M, Park K, Kim H-N. Connexin26 mutations associated with nonsyndromic hearing loss. Laryngoscope. 2000;110:1535–8.
Wagner EL, Shin J-B. Mechanisms of hair cell damage and repair. Trends Neurosci. 2019;42:414–24.
Franco B, Malgrange B. Concise review: regeneration in mammalian cochlea hair cells: help from supporting cells transdifferentiation. Stem Cells. 2017;35:551–6.
Shu Y, Li W, Huang M, Quan YZ, Scheffer D, Tian C, et al. Renewed proliferation in adult mouse cochlea and regeneration of hair cells. Nat Commun. 2019;10:5530.
Sacheli R, Delacroix L, Vandenackerveken P, Nguyen L, Malgrange B. Gene transfer in inner ear cells: a challenging race. Gene Ther. 2013;20:237–47.
Akil O, Dyka F, Calvet C, Emptoz A, Lahlou G, Nouaille S, et al. Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model. Proc Natl Acad Sci USA. 2019;116:4496–501.
Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci. 2017;20:1172–9.
Deverman BE, Pravdo PL, Simpson BP, Kumar SR, Chan KY, Banerjee A, et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol. 2016;34:204–9.
Suzuki J, Hashimoto K, Xiao R, Vandenberghe LH, Liberman MC. Cochlear gene therapy with ancestral AAV in adult mice: complete transduction of inner hair cells without cochlear dysfunction. Sci Rep. 2017;7:45524.
Yoshimura H, Shibata SB, Ranum PT, Moteki H, Smith RJH. Targeted allele suppression prevents progressive hearing loss in the mature murine model of human TMC1 deafness. Mol Ther. 2019;27:681–90.
Isgrig K, McDougald DS, Zhu J, Wang HJ, Bennett J, Chien WW. AAV2.7m8 is a powerful viral vector for inner ear gene therapy. Nat Commun. 2019;10:427–427.
György B, Meijer EJ, Ivanchenko MV, Tenneson K, Emond F, Hanlon KS, et al. Gene transfer with AAV9-PHP.B rescues hearing in a mouse model of Usher syndrome 3A and transduces hair cells in a non-human primate. Mol Ther. 2019;13:1–13.
Dulon D, Papal S, Patni P, Cortese M, Vincent PF, Tertrais M, et al. Clarin-1 gene transfer rescues auditory synaptopathy in model of Usher syndrome. J Clin Investig. 2018;128:3382–401.
Pan B, Askew C, Galvin A, Heman-Ackah S, Asai Y, Indzhykulian AA, et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat Biotechnol. 2017;35:264–72.
Yu Q, Wang Y, Chang Q, Wang J, Gong S, Li H, et al. Virally expressed connexin26 restores gap junction function in the cochlea of conditional Gjb2 knockout mice. Gene Ther. 2014;21:71–80.
Shibata SB, Ranum PT, Moteki H, Pan B, Goodwin AT, Goodman SS, et al. RNA interference prevents autosomal-dominant hearing loss. Am J Hum Genet. 2016;98:1101–13.
Landegger LD, Pan B, Askew C, Wassmer SJ, Gluck SD, Galvin A, et al. A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat Biotechnol. 2017;35:280–4.
Ren Y, Landegger LD, Stankovic KM. Gene therapy for human sensorineural hearing loss. Front Cell Neurosci. 2019;13:323–323.
Jüttner J, Szabo A, Gross-Scherf B, Morikawa RK, Rompani SB, Hantz P, et al. Targeting neuronal and glial cell types with synthetic promoter AAVs in mice, non-human primates and humans. Nat Neurosci. 2019;22:1345–56.
Hu X, Wang J, Yao X, Xiao Q, Xue Y, Wang S, et al. Screened AAV variants permit efficient transduction access to supporting cells and hair cells. Cell Discov. 2019;5:49.
Lee J, Nist-Lund C, Solanes P, Goldberg H, Wu J, Pan B, et al. Efficient viral transduction in mouse inner ear hair cells with utricle injection and AAV9-PHP.B. Hear Res. 2020;107882.
Shu Y, Tao Y, Wang Z, Tang Y, Li H, Dai P, et al. Identification of adeno-associated viral vectors that target neonatal and adult mammalian inner ear cell subtypes. Hum Gene Ther. 2016;27:687–99.
Gu X, Chai R, Guo L, Dong B, Li W, Shu Y, et al. Transduction of adeno-associated virus vectors targeting hair cells and supporting cells in the neonatal mouse cochlea. Front Cell Neurosci. 2019;13:8.
Tao Y, Huang M, Shu Y, Ruprecht A, Wang H, Tang Y, et al. Delivery of adeno-associated virus vectors in adult mammalian inner-ear cell subtypes without auditory dysfunction. Hum Gene Ther. 2018;29:492–506.
Liu Y, Qi J, Chen X, Tang M, Chu C, Zhu W, et al. Critical role of spectrin in hearing development and deafness. Sci Adv. 2019;5:eaav7803.
Chien WW, McDougald DS, Roy S, Fitzgerald TS, Cunningham LL. Cochlear gene transfer mediated by adeno-associated virus: comparison of two surgical approaches. Laryngoscope. 2015;125:2557–64.
Bermingham NA, Hassan BA, Price SD, Vollrath MA, Ben-Arie N, Eatock RA, et al. Math1: an essential gene for the generation of inner ear hair cells. Science. 1999;284:1837–41.
Zhong L, Li B, Mah CS, Govindasamy L, Agbandje-McKenna M, Cooper M, et al. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci USA. 2008;105:7827–32.
Berns KI, Srivastava A. Next generation of adeno-associated virus vectors for gene therapy for human liver diseases. Gastroenterol Clin North Am. 2019;48:319–30.
Buning H, Srivastava A. Capsid modifications for targeting and improving the efficacy of AAV vectors. Mol Ther Methods Clin Dev. 2019;12:248–65.
van Lieshout LP, Domm JM, Rindler TN, Frost KL, Sorensen DL, Medina SJ, et al. A novel triple-mutant AAV6 capsid induces rapid and potent transgene expression in the muscle and respiratory tract of mice. Mol Ther Methods Clin Dev. 2018;9:323–9.
Li W, Asokan A, Wu Z, Van Dyke T, DiPrimio N, Johnson JS, et al. Engineering and selection of shuffled AAV genomes: a new strategy for producing targeted biological nanoparticles. Mol Ther. 2008;16:1252–60.
Koerber JT, Jang JH, Schaffer DV. DNA shuffling of adenoassociated virus yields functionally diverse viral progeny. Mol Ther. 2008;16:1703–9.
Grimm D, Lee JS, Wang L, Desai T, Akache B, Storm TA, et al. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J Virol. 2008;82:5887–911.
Choudhury SR, Fitzpatrick Z, Harris AF, Maitland SA, Ferreira JS, Zhang Y, et al. In vivo selection yields AAV-B1 capsid for central nervous system and muscle gene therapy. Mol Ther. 2016;24:1247–57.
Mathelier A, Fornes O, Arenillas DJ, Chen C-y, Denay G, Lee J, et al. JASPAR 2016: a major expansion and update of the openaccess database of transcription factor binding profiles. Nucleic Acids Res. 2015;44:D110–5.
Hai T, Cao C, Shang H, Guo W, Mu Y, Yang S, et al. Pilot study of large-scale production of mutant pigs by ENU mutagenesis. Elife. 2017;6:e26248.
Hao QQ, Li L, Chen W, Jiang QQ, Ji F, Sun W, et al. Key genes and pathways associated with inner ear malformation in SOX10 (p.R109W) mutation pigs. Front Mol Neurosci. 2018;11:181.
Chang Q, Wang J, Li Q, Kim Y, Zhou B, Wang Y, et al. Virally mediated Kcnq1 gene replacement therapy in the immature scala media restores hearing in a mouse model of human Jervell and Lange-Nielsen deafness syndrome. EMBO Mol Med. 2015;7:1077–86.
Kim MA, Cho HJ, Bae SH, Lee B, Oh SK, Kwon TJ, et al. Methionine sulfoxide reductase B3-targeted In utero gene therapy rescues hearing function in a mouse model of congenital sensorineural hearing loss. Antioxid Redox Signal. 2016;24:590–602.
Kim MA, Kim SH, Ryu N, Ma JH, Kim YR, Jung J, et al. Gene therapy for hereditary hearing loss by SLC26A4 mutations in mice reveals distinct functional roles of pendrin in normal hearing. Theranostics. 2019;9:7184–99.
Iizuka T, Kamiya K, Gotoh S, Sugitani Y, Suzuki M, Noda T, et al. Perinatal Gjb2 gene transfer rescues hearing in a mouse model of hereditary deafness. Hum Mol Genet. 2015;13:13.
Isgrig K, Shteamer JW, Belyantseva IA, Drummond MC, Fitzgerald TS, Vijayakumar S, et al. Gene therapy restores balance and auditory functions in a mouse model of usher syndrome. Mol Ther. 2017;25:780–91.
Geng R, Omar A, Gopal RS, Chen H-CD, Stepanyan R, Basch LM, et al. Modeling and preventing progressive hearing loss in Usher syndrome III. Sci Rep. 2017;7:13480.
Al-Moyed H, Cepeda AP, Jung S, Moser T, Kügler S, Reisinger E. A dual-AAV approach restores fast exocytosis and partially rescues auditory function in deaf otoferlin knock-out mice. EMBO Mol Med. 2019;11:e9396.
Askew C, Rochat C, Pan B, Asai Y, Ahmed H, Child E, et al. Tmc gene therapy restores auditory function in deaf mice. Sci Transl Med. 2015;7:295ra108.
Nist-Lund CA, Pan B, Patterson A, Asai Y, Chen T, Zhou W, et al. Improved TMC1 gene therapy restores hearing and balance in mice with genetic inner ear disorders. Nat Commun. 2019;10:236.
Gao X, Tao Y, Lamas V, Huang M, YehW-H, Pan B, et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature. 2018;553:217–21.
Simmons DD, Tong B, Schrader AD, Hornak AJ. Oncomodulin identifies different hair cell types in the mammalian inner ear. J Comp Neurol. 2010;518:3785–802.
Maison S, Liberman LD, Liberman MC Type II cochlear ganglion neurons do not drive the olivocochlear reflex: re-examination of the cochlear phenotype in peripherin knock-out mice. eNeuro. 2016;3:ENEURO.0207–16.2016.
Zheng J, Shen W, He DZ, Long KB, Madison LD, Dallos P. Prestin is the motor protein of cochlear outer hair cells. Nature. 2000;405:149–55.
McLean WJ, Yin X, Lu L, Lenz DR, McLean D, Langer R, et al. Clonal expansion of Lgr5-positive cells from mammalian cochlea and high-purity generation of sensory hair cells. Cell Rep. 2017;18:1917–29.
Bermingham-McDonogh O, Oesterle EC, Stone JS, Hume CR, Huynh HM, Hayashi T. Expression of Prox1 during mouse cochlear development. J Comp Neurol. 2006;496:172–86.
Boström M, Anderson M, Lindholm D, Park K-H, Schrott-Fischer A, Pfaller K, et al. Neural network and “ganglion” formations in vitro: a video microscopy and scanning electron microscopy study on adult cultured spiral ganglion cells. Otol Neurotol. 2007;28:1109–19.
Ranum P, Goodwin A, Yoshimura H, Kolbe D, Walls W, Koh JY, et al. Insights into the biology of hearing and deafness revealed by single-cell RNA sequencing. Cell Rep. 2019;26:3160–71.
Rio C, Dikkes P, Liberman MC, Corfas G. Glial fibrillary acidic protein expression and promoter activity in the inner ear of developing and adult mice. J Comp Neurol. 2002;442:156–62.
Ladrech S, Lenoir M, Ruel J, Puel J-L. Microtubule-associated protein 2 (MAP2) expression during synaptic plasticity in the guinea pig cochlea. Hear Res. 2003;186:85–90.
Liu H, Pecka JL, Zhang Q, Soukup GA, Beisel KW, He DZZ. Characterization of transcriptomes of cochlear inner and outer hair. Cells. 2014;34:11085–95.
Pannier S, Couloigner V, Messaddeq N, Elmaleh-Berges M, Munnich A, Romand R, et al. Activating Fgfr3 Y367C mutation causes hearing loss and inner ear defect in a mouse model of chondrodysplasia. Biochim Biophys Acta. 2009;1792:140–7.
Huang X, Liu J, Wu W, Hu P, Wang Q. Taurine enhances mouse cochlear neural stem cell transplantation via the cochlear lateral wall for replacement of degenerated spiral ganglion neurons via sonic hedgehog signaling pathway. Cell Tissue Res. 2019;378:49–57.
Parker MA, Jiang K, Kempfle JS, Mizutari K, Simmons CL, Bieber R, et al. TAK1 expression in the cochlea: a specific marker for adult supporting cells. J Assoc Res Otolaryngol. 2011;12:471–83.
Spencer RF, Shaia WT, Gleason AT, Sismanis A, Shapiro SM. Changes in calcium-binding protein expression in the auditory brainstem nuclei of the jaundiced Gunn rat. Hear Res. 2002;171:129–41.
Hosoya M, Fujioka M, Matsuda S, Ohba H, Shibata S, Nakagawa F, et al. Expression and function of Sox21 during mouse cochlea development. Neurochem Res. 2011;36:1261–9.
Shrestha BR, Chia C, Wu L, Kujawa SG, Liberman MC, Goodrich LV. Sensory neuron diversity in the inner ear is shaped by activity. Cell. 2018;174:1229–46.e17.
Li S, Mecca A, Kim J, Caprara GA, Wagner EL, Du T-T, et al. Myosin-VIIa is expressed in multiple isoforms and essential for tensioning the hair cell mechanotransduction complex. Nature Communications. 2020;11:2066.
Oesterle EC, Campbell S, Taylor RR, Forge A, Hume CR. Sox2 and JAGGED1 expression in normal and drug-damaged adult mouse inner ear. J Assoc Res Otolaryngol. 2008;9:65–89.
Steevens AR, Glatzer JC, Kellogg CC, Low WC, Santi PA, Kiernan AE. SOX2 is required for inner ear growth and cochlear nonsensory formation before sensory development. Development. 2019;146:dev170522.
Vyas P, Wu JS, Jimenez A, Glowatzki E, Fuchs PA. Characterization of transgenic mouse lines for labeling type I and type II afferent neurons in the cochlea. Sci Rep. 2019;9:5549.
Roux I, Hosie S, Johnson SL, Bahloul A, Cayet N, Nouaille S, et al. Myosin VI is required for the proper maturation and function of inner hair cell ribbon synapses. Hum Mol Genet. 2009;18:4615–28.
Nagy I, Bodmer M, Schmid S, Bodmer D. Promyelocytic leukemia zinc finger protein localizes to the cochlear outer hair cells and interacts with prestin, the outer hair cell motor protein. Hear Res. 2005;204:216–22.
Hertzano R, Puligilla C, Chan SL, Timothy C, Depireux DA, Ahmed Z, et al. CD44 is a marker for the outer pillar cells in the early postnatal mouse inner ear. J Assoc Res Otolaryngol. 2010;11:407–18.
Froud KE, Wong ACY, Cederholm JME, Klugmann M, Sandow SL, Julien J-P, et al. Type II spiral ganglion afferent neurons drive medial olivocochlear reflex suppression of the cochlear amplifier. Nat Commun. 2015;6:7115.
Pan B, Géléoc Gwenaelle S, Asai Y, Horwitz Geoffrey C, Kurima K, Ishikawa K, et al. TMC1 and TMC2 are components of the mechanotransduction channel in hair cells of the mammalian inner ear. Neuron. 2013;79:504–15.
Bermingham-McDonogh O, Oesterle EC, Stone JS, Hume CR, Huynh HM, Hayashi T. Expression of Prox1 during mouse cochlear development. J Comp Neurol. 2006;496:172–86.
Liu S, Wang Y, Lu Y, Li W, Liu W, Ma J, et al. The key transcription factor expression in the developing vestibular and auditory sensory organs: a comprehensive comparison of spatial and temporal patterns. Neural Plast. 2018;2018:7513258.
Xie D, Hu P, Xiao ZA, Wu W, Chen Y, Xia K. Subunits of voltage-gated calcium channels in murine spiral ganglion cells. Acta oto-laryngologica. 2007;127:8–12.
Zhao HB, Yu N. Distinct and gradient distributions of connexin26 and connexin30 in the cochlear sensory epithelium of guinea pigs. J Comp Neurol. 2006;499:506–18.
Yang T, Scholl ES, Pan N, Fritzsch B, Haeseleer F, Lee A. Expression and localization of CaBP Ca2+ binding proteins in the mouse cochlea. PLoS ONE. 2016;11:e0147495.
Li J, Verkman AS. Impaired hearing in mice lacking aquaporin-4 water channels. J Biol Chem. 2001;276:31233–7.
Girotto G, Vuckovic D, Buniello A, Lorente-Cánovas B, Lewis M, Gasparini P, et al. Expression and replication studies to identify new candidate genes involved in normal hearing function. PLoS ONE. 2014;9:e85352–e85352.
Scheffer DI, Shen J, Corey DP, Chen ZY. Gene expression by mouse inner ear hair cells during development. J Neurosci. 2015;35:6366–80.
Huang EJ, Liu W, Fritzsch B, Bianchi LM, Reichardt LF, Xiang M. Brn3a is a transcriptional regulator of soma size, target field innervation and axon pathfinding of inner ear sensory neurons. Development. 2001;128:2421–32.
Hickox AE, Wong ACY, Pak K, Strojny C, Ramirez M, Yates JR, et al. Global analysis of protein expression of inner ear hair. Cells. 2017;37:1320–39.
Acknowledgements
This work was supported by the National Natural Science Foundation of China, 81970878 and 31771130 (GZ), Shanghai Municipal Government, and ShanghaiTech University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lan, Y., Tao, Y., Wang, Y. et al. Recent development of AAV-based gene therapies for inner ear disorders. Gene Ther 27, 329–337 (2020). https://doi.org/10.1038/s41434-020-0155-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41434-020-0155-7
This article is cited by
-
Spotlight on gene therapy in China
Gene Therapy (2020)
