
Abstract
B cells have multiple actions on different phases of an immune reaction, mainly resulting in B and T cell-interaction (help), production of cytokines, regulation of dendritic cells and downregulation of regulatory B cells. The effectiveness of B cell depletion therapy is probably due to blockade of the antigen-presenting function of B cells, occurring very early in the setting of autoimmune reactions. B cells undergo a maturation process from stem cells during which the CD 20 antigen, which is the target of rituximab (RTX), is expressed from the stage of pre-B cells to mature and memory B cells, but not on plasma cells. During the maturation process, the cytokine B cell stimulating factor (BAFF) induces maturation of B cells and expansion of clones to produce plasma cells and eventually antibodies. The effect of RTX in GO is rather rapid, with significant improvement of the disease already 4–6 weeks after the first RTX infusion. Based on the evidence of significant lymphocytic infiltration in the orbits of patients with active GO, it is reasonable to postulate that RTX may cause depletion of B cells and block their antigen-presenting cell mechanism. Since it has been reported that serum BAFF concentrations are elevated in hyperthyroid GD patients and that BAFF is expressed on the thyrocytes of patients with either autoimmune disease or nodular goiter, the hypothesis that belimumab, an anti-BAFF monoclonal antibody, may be effective in patients with active GO his currently being tested in a randomized controlled trial.
Similar content being viewed by others


The development of immunotherapy, based on antigen-specific monoclonal antibodies, has allowed to uncover the role of different immune effectors in the pathogenesis of autoimmune disease. Over the past decade several monoclonal antibodies have been employed in both open label and randomized clinical trials in Graves’ Orbitopathy (GO) [1]. Some of these molecules have been shown to be effective or promising novel therapies for the active, moderate-severe phase of the disease. Among the available therapeutic agents, those targeting B cells or B cell actions have gained interest because of their potential efficacy in GO which sheds light on underlying novel roles of B cells in the disease pathogenesis.
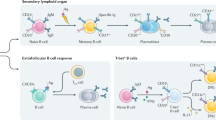
B cells, besides the well-known role as antibody-producing cells, have multiple actions on different phases in the cascade of events that regulate the progression of an immune reaction in autoimmune disease [2]. Their main action is the result of B and T-cell-interaction (help) and costimulation. But B cells also produce cytokines, mainly IL-4, IL-6, IL-10, gamma-IFN and TGF-beta and contribute to organogenesis of lymphoid organs, regulation of dendritic cells and downregulation of the production of regulatory B cells (Fig. 1). One important hypothesized role of B cells, which may explain why B cell depletion with the anti-CD 20 monoclonal antibody (rituximab; RTX) is an effective treatment in a mainly T-cell-mediated disease like GO, is a strong antigen-presenting function occurring probably very early in the setting of autoimmune reactions. In 2011 Ueki and colleagues [3] have studied an experimental Graves’ disease model (mouse) in which they have observed that B cell depletion of the mice with RTX, carried out before immunization with adenovirus expressing TSH receptor A-subunit (Ad-TSHR289), inhibits the production of autoimmunity (circulating anti-TSH receptor antibodies) and of hyperthyroidism, measured as increased circulating free thyroxine (freeT4). When RTX is applied to the mice after the first or third cycle of immunization with the TSH receptor fragment, the development of hyperthyroidism and anti-TSH receptor antibodies is not prevented. These data in experimental Graves’ disease suggest that B cells are essential in the early setting of autoimmune reactions leading to the development of clinically relevant hyperthyroidism. Very recent work from Smith and colleagues [4] has shown that thyroid antigen-reactive B cells (TPO and thyroglobulin) in patients with recent onset autoimmune thyroid disease in the peripheral blood are no longer anergic, but clearly express CD86, a marker of activation. Frequency of anergic B cells inversely correlated with circulating levels of thyroid autoantibodies. Therefore, the Authors suggested that loss of B cell anergy early in the development of autoimmunity may enable B cells to present antigen and receive T cell help.

The multiple actions of B cells
B cells undergo a maturation process within the germinal center from stem cells to mature B cells and memory B cells, from which plasma cells are derived (Fig. 2). The CD 20 antigen, which is the target of RTX, is expressed by B cells from the stage of pre-B cells to mature and memory B cells, but not on plasma cells, especially short-lived plasma cells. During the maturation process, the cytokine B cell stimulating factor (BAFF), through interaction with the BAFF receptor or TACI, induces maturation of B cells and expansion of clones to produce plasma cells and eventually antibodies. BAFF is targeted by a monoclonal antibody (belimumab) that possesses therapeutic effect in systemic lupus [5]. Both these therapeutics have been employed in the treatment of GO.

The line of maturation of B cells. RTX: rituximab; BAFF: B cell stimulating factor; Belimumab: anti-BAFF monoclonal antibody; TACI: BAFF receptor; CD-20: target of RTX
RTX is a chimeric murine/human monoclonal antibody with a human IgG1 constant region and a murine variable region binding to the CD 20 antigen on B cells. It causes rapid depletion of B cells in periphery and in lymphoid organs, likely including the thyroid [6] and the orbit [7,8,9] in Graves’ disease and GO, respectively. The current indications of RTX are rheumatoid arthritis and ANCA-related vasculitis [5]. The hypothesis that RTX might decrease circulating autoantibodies in autoimmune thyroid disease, as observed in rheumatoid arthritis and systemic lupus, has been challenged in a number of open label and controlled studies over the last few years. As reported by El Fassi et al. in 2007 [10], RTX administered to hyperthyroid Graves’ patients treated with antithyroid drugs induced remission in 40% of patients at one year when compared to none of the patients treated with antithyroid drugs alone and this effect was attributed to a specific decrease of the TSH receptor stimulating antibody subpopulation induced by RTX [11]. Other authors have described a decline of serum TSH receptor binding antibodies (TRAb) in patients with active GO after RTX therapy, whether the patients were on antithyroid drugs or euthyroid on L-T4 replacement therapy [12], suggesting again a direct effect of RTX on antibodies. Others have found that the beneficial effect of RTX on GO was not correlated to the decrease of serum TRAb [13]. A more precise picture of the effect of RTX on TRAb has been provided by the study of Vannucchi et al [14], who were not able to show a distinct decrease of circulating TSH receptor stimulating antibodies in patients with GD and GO after RTX, if compared to the decline of serum TSH receptor binding antibodies (TRAb) observed during the patients’ follow-up when they reached the euthyroid state.
The efficacy of B-cell-depleting agents in GO must then depend on other functions of B cells, other than decreased production of pathogenic antibodies. This is more evident from the studies conducted in Graves’ hyperthyroidism, which is caused by TSH receptor stimulating antibodies. The reasons for the efficacy of RTX in GO are more difficult to understand, as we do not have a disease specific immune marker: in the eye disease TRAb have been shown to correlate with its severity [15] but may not be pathogenic [16]. Consistently with what observed earlier on by Ho et al [17] and in our laboratory [18], recent data have also shown a correlation between the degree of infiltration of B (and T) cells in the orbit of patients with GO and their clinical characteristics, especially the CAS and diplopia [19]. Treatment with RTX has been shown to inactivate GO in patients with moderate-severe active disease [20]. The effect of the anti-CD20 antibody is rather rapid, with significant improvement of the disease assessed by the decrease of the CAS already 4–6 weeks after the first RTX infusion. Some inconsistencies in the clinical response in terms of efficacy of RTX have been observed in two different randomized trials, one in which RTX has been shown to be better than intravenous methylprednisolone in inactivating GO and another in which RTX efficacy was not different from placebo [21]. Post-hoc analysis of the two studies has shown that the inclusion of patients with different disease duration may explain the different therapeutic response. When RTX is administered early in the disease course seems to be very effective, whereas when used later in the disease (>12–15 months) such treatment may not be successful [22]. Based on the evidence of early, significant lymphocytic infiltration in the orbits of patients with active GO [8, 19, 23], it is reasonable to postulate that RTX, when administered early, may cause depletion of B cells and block their antigen presenting action which occurs early in the development of autoimmune reactions. Indeed, when B cells begin repopulate in the circulation, usually between 8 and 16 weeks after RTX administration depending on the dose administered, inflammation of GO does not relapse and the CAS continues to decline until definite inactivation of disease (Fig. 3). In clinical trials, GO patients who have responded successfully to RTX, have shown disease relapse in less than 7% of patients, when compared to 50% of patients treated with i.v. methylprednisolone, despite the persistence of elevated titers of TRAb and of hyperthyroidism [22]. GO is typically characterized by a single dyphasic disease course [24], an active, inflammatory progressive phase followed by orbital fibrosis and scarring. We believe that RTX acts by rapidly switching off inflammation when used early in the disease, which subsequently burns out. This is not observed in patients with systemic autoimmunity such as remitting/relapsing rheumatoid arthritis, in whom multiple subsequent infusions of RTX are often required [25].

Relationship between CD-20 + B cell depletion and the change of the Clinical Activity Score in patients with active Graves’ orbitopathy. CAS > 3: active GO
While in moderate-severe GO total peripheral T cells have not been shown to change after RTX [20], certain T cells phenotypes, specifically the IGF-1R+ T cell phenotype, which has been shown to be overexpressed in active severe GO, is directly targeted and transiently normalized after RTX [26]. These findings suggest a potentially interesting association between changes in T cell phenotypes and the clinical responses to RTX and identify a possible role for B cells in supporting the IGF-1R+ phenotype of T cells in the disease. In addition, although studies are limited to the samples of few GO patients undergoing orbital decompression surgery for either acute optic neuropathy [8] or severe proptosis [19, 23], neither B nor T cells have been detected in the orbital tissues after RTX, independently of the duration or the activity of the disease. These findings seem to support the hypothesis that early depletion of B cells would not support the B-T cell interaction that allows the development of the autoimmune pathogenic process. Indeed, immunohistochemistry of orbital tissue of patients with GO even after a very low dose RTX (100 mg) has shown depletion of both CD20 and CD3 cells, when compared to patients treated with intravenous methylprednisolone, in whom therapy did not modify the pattern of infiltration. In the orbital tissue of GO patients after acute cytokine release induced by RTX, an important degree of infiltration of CD68 + CD163 + CD1a- macrophages has been described. These cells were probably recruited as a result of the overproduction of complement anaphylatoxins and other cytokines subsequent to the acute B cell lysis induced by RTX [23]. CD163 is a marker of type 2 macrophages which, in contrast to type 1 proinflammatory macrophages generally involved in antigen presentation, are actively initiating phagocytosis. We therefore hypothesize that B cell depletion with RTX in GO may activate at the orbital level a sequence of reactions following B cell lysis characterized by recruitment of phagocytes and depletion of all inflammatory cells from the orbit. After several weeks (4 to 16), with the return of B cells in the periphery (and the orbit), the ongoing development of intraorbital conditions of fibrosis may prevent the reactivation of GO, despite the reconstitution of the previous autoimmune conditions.
The process of maturation from transitional B cells to mature B cells (Fig. 2) is stimulated by the increased production of B-lymphocyte activating factor (BAFF, also called BLyS) and subsequent formation of plasma cells [27]. BAFF is a cytokine that belongs to the tumor necrosis factor (TNF) family and is expressed on neutrophils, monocytes, dendritic cells, lymphoid stromal cells and malignant B cells. BAFF binds to three receptors called BAFF receptor (BR3), expressed on B cell, transmembrane activator and CAML interactor (TACI), expressed on mature B cell, plasma cells, and a subset of activated T cells and B cell maturation antigen called BCMA, expressed only on B cells [28]. Serum BAFF concentrations have been found to be elevated in systemic autoimmune diseases including systemic lupus erythematosus and rheumatoid arthritis [29]. A possible role of BAFF in autoimmune hyperthyroidism has been suggested by the observation that blockade of BAFF by a BAFF-specific receptor-Fc in a murine model of Graves’ disease reduced the resulting hyperthyroidism [30]. Fabris and colleagues have detected slightly elevated serum BAFF concentrations in patients with thyroid autoimmune disease, more markedly in patients with GD compared to Hashimoto’s thyroiditis (HT) [31]. Elevated serum BAFF concentrations have been reported in hyperthyroid GD patients but not in patients with GO, even during the active phase of the disease [32]. Of interest, Campi et al. [33] have recently shown that BAFF and BAFF-R are expressed on the thyrocytes derived from patients with either autoimmune disease or nodular goiter, in addition to the expected expression of BAFF and its receptor on the infiltrating immune cells of GD and HT.
Belimumab, a fully humanized IgG1 monoclonal antibody directed against BAFF, is a biological treatment approved for patients with systemic lupus, in whom it has been shown to have beneficial effects by randomized trials involving autoantibody-positive patients with persistent disease activity, after standard immunosuppressive treatment. Belimumab inhibits B cells stimulation by blocking BAFF, thus reducing disease activity [5]. A potential beneficial role of belimumab in human GD and GO is based on the B-cell driven pathogenesis of Graves’ hyperthyroidism. Such hypothesis is currently challenged in a randomized controlled trial conducted in our laboratory, in which patients with GD with detectable serum TRAb and associated active moderate-severe GO are treated with intravenous belimumab or methylprednisolone, currently considered first line treatment. An interim analysis of the trial results of this study will become available in 2019.
References
Campi I, Vannucchi G, Salvi M. Therapy of endocrine disease: endocrine dilemma: management of Graves’ orbitopathy. Eur J Endocrinol. 2016;175:R117–33.
Yanaba K, Bouaziz JD, Matsushita T, Magro CM, St Clair EW, Tedder TF. B-lymphocyte contributions to human autoimmune disease. Immunol Rev. 2008;223:284–99.
Ueki I, Abiru N, Kobayashi M, Nakahara M, Ichikawa T, Eguchi K, et al. B cell-targeted therapy with anti-CD20 monoclonal antibody in a mouse model of Graves’ hyperthyroidism. Clin Exp Immunol. 2011;163:309–17.
Smith MJ, Rihanek M, Coleman BM, Gottlieb PA, Sarapura VD, Cambier JC. Activation of thyroid antigen-reactive B cells in recent onset autoimmune thyroid disease patients. J Autoimmun. 2018;89:82–9.
Dorner T, Lipsky PE. Beyond pan-B-cell-directed therapy - new avenues and insights into the pathogenesis of SLE. Nat Rev Rheumatol. 2016;12:645–57.
El Fassi D, Clemmensen O, Nielsen CH, Silkiss RZ, Hegedüs L. Evidence of intrathyroidal B-lymphocyte depletion after rituximab therapy in a patient with Graves’ disease. J Clin Endocrinol Metab. 2007;92:3762–3.
Salvi M, Vannucchi G, Campi I, Rossi S, Bonara P, Sbrozzi F, et al. Efficacy of rituximab treatment for thyroid-associated ophthalmopathy as a result of intraorbital B-cell depletion in one patient unresponsive to steroid immunosuppression. Eur J Endocrinol. 2006;154:511–7.
Khanna D, Chong KK, Afifiyan NF, Hwang CJ, Lee DK, Garneau HC, et al. Rituximab treatment of patients with severe, corticosteroid-resistant thyroid-associated ophthalmopathy. Ophthalmology. 2010;117:133–9.
Nielsen JF, El Fassi D, Nielsen CH, Hegedüs L, Lauer SA, Silkiss RZ, et al. Evidence of orbital B and T cell depletion after rituximab therapy in Graves’ ophthalmopathy. Acta Ophthalmol. 2009;87:927–9.
El Fassi D, Nielsen CH, Bonnema SJ, Hasselbalch HC, Hegedüs L. B lymphocyte depletion with the monoclonal antibody rituximab in Graves’ disease: a controlled pilot study. J Clin Endocrinol Metab. 2007;92:1769–72.
El Fassi D, Banga JP, Gilbert JA, Padoa C, Hegedüs L, Nielsen CH. Treatment of Graves’ disease with rituximab specifically reduces the production of thyroid stimulating autoantibodies. Clin Immunol. 2009;130:252–8.
Mitchell AL, Gan EH, Morris M, Johnson K, Neoh C, Dickinson AJ, et al. The effect of B cell depletion therapy on anti-TSH receptor antibodies and clinical outcome in glucocorticoid-refractory Graves’ orbitopathy. Clin Endocrinol (Oxf). 2013;79:437–42.
Erdei A, Paragh G, Kovacs P, Karanyi Z, Berenyi E, Galuska L, et al. Rapid response to and long-term effectiveness of anti-CD20 antibody in conventional therapy resistant Graves’ orbitopathy: a five-year follow-up study. Autoimmunity. 2014;47:548–55.
Vannucchi G, Campi I, Bonomi M, Covelli D, Dazzi D, Currò N, et al. Rituximab treatment in patients with active Graves’ orbitopathy: effects on proinflammatory and humoral immune reactions. Clin Exp Immunol. 2010;161:436–43.
Eckstein AK, Plicht M, Lax H, Neuhäuser M, Mann K, Lederbogen S, et al. Thyrotropin receptor autoantibodies are independent risk factors for Graves’ ophthalmopathy and help to predict severity and outcome of the disease. J Clin Endocrinol Metab. 2006;91:3464–70.
Wiersinga WM. Advances in treatment of active, moderate-to-severe Graves’ ophthalmopathy. Lancet Diabetes Endocrinol. 2017;5:134–42.
Ho VH, Chevez-Barrios P, Jorgensen JL, Silkiss RZ, Esmaeli B. Receptor expression in orbital inflammatory syndromes and implications for targeted therapy. Tissue Antigens. 2007;70:105–9.
Salvi M, Vannucchi G, Campi I, Currò N, Simonetta S, Covelli D, et al. Rituximab treatment in a patient with severe thyroid-associated ophthalmopathy: effects on orbital lymphocytic infiltrates. Clin Immunol. 2009;131:360–5.
Rotondo Dottore G, Torregrossa L, Caturegli P, Ionni I, Sframeli A, Sabini E, et al. Association of T and B cells infiltrating orbital tissues with clinical features of Graves' orbitopathy. JAMA Ophthalmol. 2018;136:613–9.
Salvi M, Vannucchi G, Currò N, Campi I, Covelli D, Dazzi D, et al. Efficacy of B-cell targeted therapy with rituximab in patients with active moderate to severe Graves’ orbitopathy: a randomized controlled study. J Clin Endocrinol Metab. 2015;100:422–31.
Stan MN, Garrity JA, Carranza Leon BG, Prabin T, Bradley EA, Bahn RS. Randomized controlled trial of rituximab in patients with Graves’ orbitopathy. J Clin Endocrinol Metab. 2015;100:432–41.
Stan MN, Salvi M. Management of endocrine disease: rituximab therapy for Graves’ orbitopathy - lessons from randomized control trials. Eur J Endocrinol. 2017;176:R101–109.
Salvi M, Vannucchi G, Currò N, Introna M, Rossi S, Bonara P, et al. Small dose of rituximab for Graves' orbitopathy: new insights into the mechanism of action. Arch Ophthalmol. 2012;130:122–4.
Bartley GB. Rundle and his curve. Arch Ophthalmol. 2011;129:356–8.
Henry J, Gottenberg JE, Rouanet S, Pavy S, Sellam J, Tubach F, et al. Doses of rituximab for retreatment in rheumatoid arthritis: influence on maintenance and risk of serious infection. Rheumatol (Oxf). 2018;57:538–47.
McCoy AN, Kim DS, Gillespie EF, Atkins SJ, Smith TJ, Douglas RS. Rituximab (Rituxan) therapy for severe thyroid-associated ophthalmopathy diminishes IGF-1R(+) T cells. J Clin Endocrinol Metab. 2014;99:E1294–9.
Rowland SL, Leahy KF, Halverson R, Torres RM, Pelanda R. BAFF receptor signaling aids the differentiation of immature B cells into transitional B cells following tonic BCR signaling. J Immunol. 2010;185:4570–81.
Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404:995–9.
Pers JO, Daridon C, Devauchelle V, Jousse S, Saraux A, Jamin C, et al. BAFF overexpression is associated with autoantibody production in autoimmune diseases. Ann N Y Acad Sci. 2005;1050:34–9.
Gilbert JA, Kalled SL, Moorhead J, HessDM, Rennert P, Li Z, et al. Treatment of autoimmune hyperthyroidism in a murine model of Graves' disease with tumor necrosis factor-family ligand inhibitors suggests a key role for B cell activating factor in disease pathology. Endocrinology. 2006;147:4561–8.
Fabris M, Grimaldi F, Villalta D, Picierno A, Fabro C, Bolzan M, et al. BLyS and April serum levels in patients with autoimmune thyroid diseases. Autoimmun Rev. 2010;9:165–9.
Vannucchi G, Covelli D, Currò N, Dazzi D, Maffini A, Campi I, et al. Serum BAFF concentrations in patients with Graves’ disease and orbitopathy before and after immunosuppressive therapy. J Clin Endocrinol Metab. 2012;97:E755–9.
Campi I, Tosi D, Rossi S, Vannucchi G, Covelli D, Colombo F, et al. B Cell Activating Factor (BAFF) and BAFF Receptor Expression in Autoimmune and Nonautoimmune Thyroid Diseases. Thyroid. 2015;25:1043–9.
Funding
This work was in part supported by Fondazione IRCCS Cà Granda, Ospedale Maggiore Policlinico, Milan Italy to Mario Salvi (mario@mariosalvinet.it).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
MS has a consulting agreement with Immunovant Inc., USA. The remaining author declares that she has no conflict of interest.
Rights and permissions
About this article

Cite this article
Salvi, M., Covelli, D. B cells in Graves’ Orbitopathy: more than just a source of antibodies?. Eye 33, 230–234 (2019). https://doi.org/10.1038/s41433-018-0285-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-018-0285-y
This article is cited by
-
New insights into the pathogenesis and nonsurgical management of Graves orbitopathy
Nature Reviews Endocrinology (2020)
