
Abstract
Nystagmus (involuntary, rhythmical eye movements) can arise due to sensory eye defects, in association with neurological disorders or as an isolated condition. We identified a family with early onset nystagmus and additional neurological features carrying a partial duplication of FGF14, a gene associated with spinocerebellar ataxia type 27 (SCA27) and episodic ataxia. Detailed eye movement analysis revealed oculomotor anomalies strikingly similar to those reported in a previously described four-generation family with early onset nystagmus and linkage to a region on chromosome 13q31.3-q33.1 (NYS4). Since FGF14 lies within NYS4, we revisited the original pedigree using whole genome sequencing, identifying a 161 kb heterozygous deletion disrupting FGF14 and ITGBL1 in the affected individuals, suggesting an FGF14-related condition. Therefore, our study reveals the genetic variant underlying NYS4, expands the spectrum of pathogenic FGF14 variants, and highlights the importance of screening FGF14 in apparently isolated early onset nystagmus.
Similar content being viewed by others
Introduction
Congenital and early onset nystagmus (involuntary, repetitive oscillation of the eyes) typically manifests within the first months of life. It can be apparently isolated, associated with visual deficits, or seen in the context of numerous neurological disorders. Given the genetic and clinical heterogeneity of these conditions, detailed visual and neurological phenotyping, with analysis of supranuclear eye movements, can direct clinicians towards the underlying genetic causes [1, 2]. However, typical patterns of clinical features suggesting an underlying cause, such as those observed in Infantile Nystagmus Syndrome (INS) or cerebellar-type nystagmus, are not always present [3]. Whole-scale genetic testing is now assisting in diagnosing complex disorders such as nystagmus and, as described here, redefining phenotypes associated with individual gene-related conditions.
Here, we describe a father and son with nystagmus, early onset tremor, and motor difficulties, including mild ataxia. Array-CGH revealed that both individuals carry a partial duplication of FGF14 (Fibroblast Growth Factor 14, OMIM: 601515). Heterozygous FGF14 variants are associated with spinocerebellar ataxia type 27 (SCA27) [4] and episodic ataxia (EA) [5], although some individuals display milder phenotypes, including tremor without ataxia [5] or nystagmus with occasional episodes of vertigo and incoordination [6]. Detailed eye movement analysis revealed oculomotor anomalies strikingly similar to those described in a large dominant pedigree with linkage to a locus on chromosome 13q31.3-q33.1 (NYS4, OMIM: 193003) [7, 8], containing FGF14. Herein, we revisited the original NYS4 pedigree and identified a heterozygous deletion disrupting FGF14 and ITGBL1 (Integrin Subunit Beta Like 1, OMIM: 604234), segregating with the disorder. Therefore, this study determines the genetic variant underlying NYS4 and highlights the importance of FGF14 structural variants in milder forms of SCA27, including apparently isolated childhood nystagmus.
Cases and methods
Families 1 and 2 were recruited to a national ‘Genetics of Eye and Brain Anomalies study’ (REC 04/Q0104/129). Informed consent was obtained according to the tenets of the Declaration of Helsinki.
Family 1: Copy Number Variant (CNV) screening was performed using a 60-mer oligo-array (8x60K International Standard Cytogenomic Array [ISCA] Consortium configuration [Oxford Gene Technology, Oxford, UK]). Paternal DNA was sequenced with an Illumina HiSeq and SureSelect Ataxia Panel v1 including FGF14 (Agilent Technologies, Santa Clara, CA, USA).
Family 2: Whole genome sequencing (WGS) was performed using paired-end, 2 × 150, and 30x coverage with an Illumina NovaSeq 6000 (Theragen Bio, Republic of Korea). The presence of sequence variants in diagnostic ataxia or nystagmus genes was assessed (PanelApp panels “Hereditary ataxia and cerebellar anomalies - childhood onset” v6.28, “Albinism or congenital nystagmus” v1.5, “Infantile nystagmus” v1.3; https://panelapp.genomicsengland.co.uk/). Structural variants were identified using bbmap (https://sourceforge.net/projects/bbmap/). Breakpoints were identified from bbmap-aligned files using the GRIDSS package [9] and validated by PCR and Sanger sequencing.
Both CNVs were evaluated according to the ACMG guidelines [10] using the ClinGen CNV Interpretation Calculator (https://cnvcalc.clinicalgenome.org/cnvcalc/).
Results
Family 1
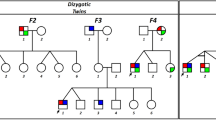
An 8-year-old boy (II.3, Fig. 1A) was referred to the eye clinic with apparently isolated nystagmus since age 4 years. History and clinical examination revealed that he had mild developmental delay and had started walking after age 2 years. His visual acuity was within normal range (logMAR < 0.18 either eye). He had vertical upbeat nystagmus in primary position, horizontal gaze-evoked nystagmus in side gazes and horizontal rebound nystagmus. Eye movement recordings showed that horizontal and upward smooth pursuits were absent, but downward smooth pursuits were present with reduced gain. His electroretinogram (ERG), visual evoked potentials (VEPs), and cranial magnetic resonance imaging (MRI) were normal. Subsequent neurological examination identified bilateral intention tremor, mild dysmetria, dysdiadochokinesis, and difficulties with heel-to-toe walking. He also had behavioural issues, including mood disorder and aggressiveness (Table 1, Supplemental Material).

A Pedigree of family 1. The proband (II.3) is the third child of nonconsanguineous parents. Black-filled symbols indicate a SCA27 phenotype. B Pedigree of family 2, with individuals numbered according to recruitment order. Black-filled symbols represent individuals with eye movement anomalies. Individuals III.63 and III.64 were assessed through a video call. Question marks indicate individuals with affected status unknown. The obligate carrier status of individual II.10 is indicated by a black dot.
His father (I.1) had poor balance, fine motor difficulties, and mood disorder. He had a history of tremor since childhood, initially attributed to asthma medication. He displayed mild left beating nystagmus in primary position, and eye movement recordings showed subtly asymmetric horizontal smooth pursuits. This was only evident on eye tracking with normal smooth pursuit response when moving the eyes to the left, but mildly reduced gain (the ratio of eye velocity to target velocity) when moving the eyes to the right. Neurological examination showed similar findings to the proband, including mild ataxia and mild intention tremor. His cranial MRI was normal (Table 1). The proband’s two sisters and mother had no medical problems.
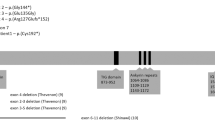
Array-CGH identified a partial FGF14 duplication in both I.1 and II.3 between ~280 kb (chr13:102,535,482-102,815,349, hg19) and ~532 kb (chr13:102,379,344-102,911,282, hg19), which was absent from ClinVar (August 2022) and DECIPHER (April 15th 2022 release). The two main isoforms of FGF14, 1A (NM_004115) and 1B (NM_175929), differ with respect to their first exon, with the minimum coordinates of the duplication encompassing at least exon 1 of isoform 1A (Fig. 2A). Read depth analysis of next-generation sequencing data from the father and seven normal controls suggests that exons 2–3 are also included in the duplication. If the duplication is in tandem, this would potentially lead to a frameshift in isoform 1B. Given that FGF14 is a haploinsufficient gene, the CNV would therefore be classified as pathogenic [10]. Sequencing data confirmed the absence of pathogenic FGF14 single nucleotide variants (SNVs) in the father.

A UCSC schematic (GRCh37, hg19) showing ITGBL1 and FGF14. The blue bar indicates the region spanned by the duplication (family 1); the thicker region of the bar shows the minimum duplicated interval. The red bar indicates the region spanned by the deletion (family 2). B Sequence chromatogram showing the breakpoints of the deletion identified in family 2. The deleted region overlaps with 4/4 ITGBL1 isoforms (including exons 8–11 in isoform 1, exons 7–10 in isoforms 2 and 3, and exons 7–11 in isoform 4) and the last two exons of FGF14-1A/1B. The 5′ boundary maps to an intronic region of ITGBL1, 114 bp from the nearest exon. The 3′ boundary maps to FGF14 intron 3. The sequence GTTT is present at both ends of the CNV and therefore cannot be definitively ascribed to either side of the breakpoint. C Schematic of the two FGF14 isoforms 1A and 1B indicating the location of structural and sequence variants identified in this study or previously reported in cases with SCA27/EA (see Supplemental Material for references). Sequence variants refer to FGF14-1B (NM_175929). Horizontal lines indicate the FGF14 exons affected by structural variants. Arrows indicate variants extending to genes adjacent to FGF14, dashed bars indicate the exons affected by the maximum coordinates of the CNV. Note that the bars do not indicate the position of the breakpoints.
Family 2
The NYS4 pedigree [7, 8] now consists of 17 affected individuals with eye movement anomalies (Fig. 1B). These include nystagmus (gaze-evoked, upbeat and rebound), poor or absent smooth pursuit, and hyperactive vestibulo-ocular reflex. II.16 and III.29 also manifested ataxia, while II.6, II.8, and III.35 had balance problems. II.10 and III.34 reported dizzy spells and mild coordination problems, respectively, without nystagmus. Strabismus and seizures were variably present. Clinical features are summarised in Table 1.
WGS of III.63 and III.64 did not detect pathogenic SNVs in known nystagmus or ataxia genes. However, a 161 kb heterozygous deletion within the NYS4 interval was identified in both individuals (chr13:102,250,764-102,412,039, hg19), encompassing 2 exons of FGF14 and 4–5 exons of ITGBL1 (depending on isoform) (Fig. 2A, B). This CNV was also absent from ClinVar (August 2022) and DECIPHER (April 15th, 2022 release). Segregation analysis by PCR showed the deletion was present in 12/12 affected and 0/9 unaffected individuals (Table 1). The deletion was classified as pathogenic according to the ACMG guidelines [10].
Discussion
We identified FGF14 structural variants in two families with early onset nystagmus and variable neurological and behavioural features: a partial duplication of FGF14 in a two-generation family and a heterozygous 161 kb deletion disrupting FGF14 and ITGBL1 in a previously described four-generation pedigree. These data finally elucidate the genetic variant underlying NYS4, a locus previously linked to the vestibulocerebellar condition described in the latter family.
FGF14 encodes an intracellular fibroblast growth factor involved in multiple neuronal processes, including channel gating and neuronal excitability [11]. Individuals with pathogenic FGF14 variants manifest EA or develop SCA27, a progressive cerebellar ataxia frequently presenting with nystagmus, tremor, dysarthria, limb ataxia, and variably associated with psychiatric symptoms and cognitive impairment. Eighteen pathogenic variants have been reported to date, including six heterozygous deletions [12,13,14,15,16,17], three of which overlap that of family 2 (Fig. 2C). While translocations and deletions are likely to cause functional haploinsufficiency, the effect of duplications is harder to predict. The variant in family 1 is the first report of a partial FGF14 duplication and affects between one and three exons. Depending on the localisation and orientation of the duplicated fragment, this variant could alter the production, folding, localisation and/or function of the protein.
SCA27 is characterised by early onset and slow progression (ataxia onset: 23.7 ± 16.7 years), with only 13.8% of patients developing severe gait impairment [18]. In family 2, nystagmus was the most frequent and consistent feature, while balance problems were more variably present. Of note, four of five affected members exhibiting unsteadiness or ataxia were age ≥30 years at their last examination, whereas those not exhibiting ataxia/balance problems were mostly younger when examined [8]. Therefore, young age of assessment together with the variable presentation of ataxic features may account for the absence of gait impairment among family 2 carriers of the FGF14 deletion.
Phenotypic intra- and inter-familial variability is a hallmark of FGF14 variants [5, 18]. Family 2 expands this variability to include isolated nystagmus and milder clinical features. While III.63 had early onset nystagmus diagnosed by the age of three, her brother III.64 was initially reported as unaffected. Re-examination of III.64 on the basis of our genetic findings revealed a similar, but far more subtle, pattern of eye movement anomalies including horizontal gaze-evoked nystagmus, saccadic pursuit, and dysmetric saccades. Similarly, the affected status of II.10 was originally unassigned as she exhibited dizzy spells without nystagmus. While DNA was unavailable, the inheritance pattern of the deletion indicates that she is an obligate carrier, suggesting her phenotype represents an extremely mild form of SCA27. Therefore, family 2 supports an emerging model whereby mild phenotypes, including apparently isolated nystagmus, can result from variants in genes associated with ataxia [19].
Furthermore, this study highlights how detailed characterisation of oculomotor anomalies within a broader movement disorder can provide insights into the genetic basis of conditions such as SCA27. Early onset nystagmus with minimal or absent tremor and ataxia could be mistaken for other forms of nystagmus seen in infancy. In our families, the oculomotor pattern is mainly characterised by vertical nystagmus and horizontal gaze-evoked nystagmus with decelerating slow phases, which would be indicative of neurological nystagmus [20]. This supports some of the previous descriptions for FGF14-related conditions where details of eye movements are mentioned [12, 14]. However, since such detailed eye movement evaluation is rarely possible in routine clinical practice, particularly in children, we recommend the inclusion of FGF14 on gene panels for childhood nystagmus.
In conclusion, our study identifies the genetic basis of NYS4, expands the spectrum of FGF14 variants, refines the phenotypes of the associated oculomotor anomalies, and demonstrates the value of screening FGF14 in children with apparently isolated early onset nystagmus.
Data availability
The two variants described in this study have been submitted to the ClinVar repository (SCV002570104, SCV002570105).
References
Clark R, Blundell J, Dunn MJ, Erichsen JT, Giardini ME, Gottlob I, et al. The potential and value of objective eye tracking in the ophthalmology clinic. Eye. 2019;33:1200–2.
Osborne D, Theodorou M, Lee H, Ranger M, Hedley-Lewis M, Shawkat F, et al. Supranuclear eye movements and nystagmus in children: A review of the literature and guide to clinical examination, interpretation of findings and age-appropriate norms. Eye. 2019;33:261–73.
Self JE, Dunn MJ, Erichsen JT, Gottlob I, Griffiths HJ, Harris C, et al. Management of nystagmus in children: a review of the literature and current practice in UK specialist services. Eye. 2020;34:1515–34.
van Swieten JC, Brusse E, de Graaf BM, Krieger E, van de Graaf R, de Koning I, et al. A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebellar ataxia [corrected]. Am J Hum Genet. 2003;72:191–9.
Piarroux J, Riant F, Humbertclaude V, Remerand G, Hadjadj J, Rejou F, et al. FGF14-related episodic ataxia: delineating the phenotype of Episodic Ataxia type 9. Ann Clin Transl Neurol. 2020;7:565–72.
Choquet K, La Piana R, Brais B. A novel frameshift mutation in FGF14 causes an autosomal dominant episodic ataxia. Neurogenetics 2015;16:233–6.
Harris CM, Walker J, Shawkat F, Wilson J, Russell-Eggitt I. Eye movements in a familial vestibulocerebellar disorder. Neuropediatrics 1993;24:117–22.
Ragge NK, Hartley C, Dearlove AM, Walker J, Russell-Eggitt I, Harris CM. Familial vestibulocerebellar disorder maps to chromosome 13q31-q33: a new nystagmus locus. J Med Genet. 2003;40:37–41.
Cameron DL, Schröder J, Penington JS, Do H, Molania R, Dobrovic A, et al. GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res. 2017;27:2050–60.
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2020;22:245–57.
Di ReJ, Wadsworth PA, Laezza F. Intracellular Fibroblast Growth Factor 14: Emerging risk factor for brain disorders. Front Cell Neurosci. 2017;11:103.
Tucker ME, Kalb FM, Escobar LF Infant Spinocerebellar Ataxia Type 27: Early Presentation Due To a 13q33.1 Microdeletion Involving the FGF14 Gene. J Genet Syndr Gene Ther. 2013;4:1–3.
Coebergh JA, Fransen van de Putte DE, Snoeck IN, Ruivenkamp C, van Haeringen A, Smit LM. A new variable phenotype in spinocerebellar ataxia 27 (SCA 27) caused by a deletion in the FGF14 gene. Eur J Paediatr Neurol. 2014;18:413–5.
Planes M, Rooryck C, Vuillaume ML, Besnard L, Bouron J, Lacombe D, et al. SCA27 is a cause of early-onset ataxia and developmental delay. Eur J Paediatr Neurol. 2015;19:271–3.
Paucar M, Lundin J, Alshammari T, Bergendal Å, Lindefeldt M, Alshammari M, et al. Broader phenotypic traits and widespread brain hypometabolism in spinocerebellar ataxia 27. J Intern Med. 2020;288:103–15.
Amado A, Blanco MO, Repáraz-Andrade A. Spinocerebellar Ataxia 27: clinical phenotype of twin sisters with FGF14 deletion. Neuropediatrics 2017;48:131.
Zech M, Boesch S, Škorvánek M, Necpál J, Švantnerová J, Wagner M, et al. Clinically relevant copy-number variants in exome sequencing data of patients with dystonia. Parkinsonism Relat Disord. 2021;84:129–34.
Groth CL, Berman BD. Spinocerebellar Ataxia 27: a review and characterization of an evolving phenotype. Tremor Other Hyperkinet Mov. 2018;8:534.
Self J, Mercer C, Boon EM, Murugavel M, Shawkat F, Hammans S, et al. Infantile nystagmus and late onset ataxia associated with a CACNA1A mutation in the intracellular loop between s4 and s5 of domain 3. Eye. 2009;23:2251–5.
Casteels I, Harris CM, Shawkat F, Taylor D. Nystagmus in infancy. Br J Ophthalmol. 1992;76:434–7.
Acknowledgements
We would like to thank the families for their participation in our study. We also thank Dr. Richard Holt for the support in the preparation of the manuscript, Dr. Lidiya Talbot and the West Midlands Regional Genetics Service for laboratory and administrative support.
Funding
This work was supported by grants from Baillie Gifford; Microphthalmia, Anophthalmia, and Coloboma Support (MACS) (www.macs.org.uk); Oxford Brookes University Health Innovation Fund (HEIF) and the Gift of Sight Charity.
Author information
Authors and Affiliations
Contributions
JES and NKR designed the study. FC, DO, JES and NKR wrote the manuscript. FC, SC and EJC performed data generation, analysis and interpretation. NKR, DO, MJD, JES and CMH performed clinical examinations of the families. DAB carried out research coordination. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The families included in this study were recruited to a national ‘Genetics of Eye and Brain Anomalies study’ (approved by the UK Regional Ethics Committee Cambridge-East, REC 04/Q0104/129). Informed consent was obtained according to the tenets of the Declaration of Helsinki.
Informed consent
Informed consent was obtained according to the tenets of the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ceroni, F., Osborne, D., Clokie, S. et al. Analysis of Fibroblast Growth Factor 14 (FGF14) structural variants reveals the genetic basis of the early onset nystagmus locus NYS4 and variable ataxia. Eur J Hum Genet 31, 353–359 (2023). https://doi.org/10.1038/s41431-022-01197-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-022-01197-5
This article is cited by
-
2023 in the European Journal of Human Genetics
European Journal of Human Genetics (2024)
-
An Update on the Adult-Onset Hereditary Cerebellar Ataxias: Novel Genetic Causes and New Diagnostic Approaches
The Cerebellum (2024)
-
Genes=disease (?)
European Journal of Human Genetics (2023)
