
Abstract
Hereditary cancer risk syndromes are caused by germline variants, commonly in tumor suppressor genes. Most studies on hereditary cancer have been conducted in white populations. We report the largest study in Brazilian individuals with multiple ethnicities. We genotyped 1682 individuals from all country regions with Next-generation sequencing (NGS) panels. Most were women with a personal/family history of cancer, mostly breast and ovarian. We identified 321 pathogenic/likely pathogenic (P/LP) variants in 305 people (18.1%) distributed among 32 genes. Most were on BRCA1 and BRCA2 (129 patients, 26.2% and 14.3% of all P/LP, respectively), MUTYH (42 monoallelic patients, 13.1%), PALB2 (25, 7.8%), Lynch syndrome genes (17, 5.3%), and TP53 (17, 5.3%). Transheterozygosity prevalence in our sample was 0.89% (15/1682). BRCA1/BRCA2 double heterozygosity rate was 0.78% (1/129) for BRCA variants carriers and 0.06% (1/1682) overall. We evaluated the performance of the genetic testing criteria by NCCN and the Brazilian National Health Agency (ANS). The inclusion criteria currently used in Brazil fail to identify 17%–25% of carriers of P/LP variants in hereditary cancer genes. Our results add knowledge on the Brazilian spectrum of cancer risk germline variants, demonstrate that large multigene panels have high positivity rates, and indicate that Brazilian inclusion criteria for genetic testing should be improved.
Similar content being viewed by others

Introduction
Hereditary cancer risk syndromes are a group of disorders caused by germline pathogenic variants in a growing number of genes. They are the main predisposing factor in about 5–10% of all diagnosed cancers. Accurate genetic diagnosis in patients with hereditary cancer risk syndromes may reduce morbidity and mortality by allowing the adoption of specific preventative and risk reduction measures. Additionally, family members at risk can be tested and counseled, extending the clinical benefits to many individuals. Currently, there are over 30 hereditary cancer syndromes already described, the most prevalent being hereditary breast and ovarian cancer (HBOC) and Lynch syndromes. Most syndromes are inherited in an autosomal-dominant manner, with variable penetrance, and are caused by mutations in oncogenes or tumor suppressor genes [1].
Genetic diagnosis of these syndromes has been simplified using NGS-based multigene panels in clinical practice. Although extensive studies have been published on the diagnostic utility of such panels, these are mostly restricted to white populations. This study aimed to describe the frequency and type of germline pathogenic variants in cancer susceptibility genes in individuals referred for testing using hereditary cancer syndrome NGS panels in the genetically admixed Brazilian population.
Materials and methods
The study included 1682 Brazilian individuals who received a multi-gene NGS panel for hereditary cancer risk in a CAP (College of American Pathologists)-accredited laboratory of the Albert Einstein Israeli Hospital (Brazil) between July 2016 and July 2019. The patients were referred to molecular testing by clinicians in private practice and no specific clinical criteria had to be met for referral, although most patients were females with a personal or family history of breast and ovarian cancer (see Results).
After the genetic assessment, we retrospectively classified individuals according to the USA National Comprehensive Cancer Network (NCCN, www.nccn.org) testing guidelines: genetic/familial high-risk assessment versions 3.2019 and 1.2020, and the Brazilian National Health Agency criteria for genetic testing for breast and ovarian cancers (ANS 2018). We first excluded the individuals without personal or family information to do this. We then selected the individuals with variants in genes related to HBOC to evaluate the sensitivity and specificity of the testing guidelines. We intended to quantify the rate of individuals with pathogenic/likely pathogenic variants missing the opportunity for genetic testing (false negatives) due to current guidelines.
This project was approved by the Research Ethics Committee of the Institute of Integral Medicine Professor Fernando Figueira (CEP-IMIP) in Recife, Pernambuco and all individuals provided written consent for multi-gene testing (protocol number 29567220.4.1001.0071).
Sequencing was performed using NGS capture panels varying from 27 genes to expanded panels (ranging between 37 and 143 genes) that covered each exon and 20 bp of intronic sequences flanking the exons. When adequate, panels also included intronic regions of interest. The complete list of genes is available in Supplementary Table 1. Genomic DNA was extracted from peripheral blood or saliva, enzymatically fragmented, and enriched by a capture method. Sequencing was performed on MiSeq or Next-Seq 550 instruments (Illumina, San Diego, CA) using MiSeq Reagent Kit v2 (300-cycles) or Next-Seq 550 High-throughput kit (300 cycles) with >99% coverage at a minimum 50X depth.
Bioinformatic analysis was performed using GATK 3.0 best practices. VCFs were annotated using Annovar and in-house databases. Variant classification was performed strictly following the American College of Genomics and Genetics (ACMG) 2015 guidelines for sequence variant interpretation and 2018 guideline update [2, 3], in a CAP-accredited laboratory.
All the data generated by the study is shown either in the main text or in the Supplementary Materials.
Results
Study sample characteristics and cancer histories
Between July 2016 and July 2019, we collected samples and clinical/demographic characteristics from 1682 Brazilian individuals referred to a private laboratory to undergo genotyping with a cancer hereditary risk multigenic panel (Table 1).
Among the individuals, 1557 (92.6%) were women with a mean age of 47.4 ± 12.3 (range 11–88) years, and 125 (7.4%) were men with a mean age of 51.4 ± 15.2 (range 14–91) years. Regarding ethnic ancestry, 1007 (59.9%) individuals reported unknown background, 330 (19.6%) reported having multiple ethnicities (admixed), 259 (15.4%) white, 26 (1.5%) black, 22 (1.3%) Ashkenazi Jewish, 21 (1.2%) native indigenous and 17 Asian (1.0%) (Table 1).
Personal cancer history was reported by 1119 (66.5%) individuals, with breast or ovary cancers identified in 971 individuals (57.7%); colorectal cancer in 71 (4.2%); thyroid/parathyroid in 36 (2.1%); prostate cancer in 15 (0.9%); stomach cancer in 14 (0.8%); uterus, kidney, and sarcomas in 13 each (0.8%); leukemia in 11 (0.6%); non-melanoma skin and pancreas cancers in 9 each (0.5%); and melanoma in 6 (0.4%). Other types of cancer (lung, liver, bladder, larynx etc.) were identified in 17 individuals (1.0%). Overall, 270 individuals (16.1%) did not present malignant neoplasms and for 293 individuals (17.4%) the history information was not available.
Family cancer history (considering first-, second- or third-degree relatives) was reported by 1219 (72.5%) individuals: 806 (47.9%) reported breast cancer family history; 310 (23.0%) colorectal cancers; 339 (20.2%) prostate cancer; 219 (13.0%) head and neck tumors; 172 (10.2%) lung cancer; 155 (9.2%) stomach cancer; 119 (7.1%) breast and ovarian and 48 (2.9%) ovarian cancer exclusively. No family history was reported by 153 (9.1%) individuals and 310 (18.4%) did not provide information regarding family history (Table 1, Supplementary Table 2).
Genetic findings
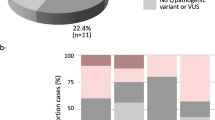
Pathogenic or likely pathogenic variants were found in 305 (18.1%) of the 1682 individuals. Additionally, 1252 variants of uncertain significance (VUS) were found in 753 (44.8%) individuals (Fig. 1A, Supplementary Table 3). The remaining 624 (37.1%) did not present any variants of clinical interest (negatives).

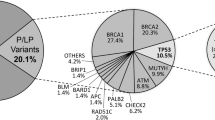
A Distribution of patients according to genetic findings. P/LP = patients with pathogenic/likely pathogenic, VUS = patients with variants of uncertain significance (N = 1682). B Distribution of P/LP variants per gene. C P/LP variants functional annotation. D Distribution of VUS per gene. Lynch syndrome genes have been grouped together in (C) and (D).
The 305 individuals collectively had 321 pathogenic/likely pathogenic variants (corresponding to 166 unique variants) in 32 genes: APC, ATM, BARD1, BRCA1, BRCA2, BRIP1, CDH1, CDK4, CHEK2, EPCAM, FANCC, MEN1, MITF, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, POLD1, PTCH2, PTEN, RAD50, RAD51, RAD51 C, RAD51D, RB1, RECQL, RET and TP53.
The genes that most commonly presented pathogenic or likely pathogenic variants were BRCA1 (84/321 = 26.2%), BRCA2 (46, 14.3%) and PALB2 (25, 7.8%). Lynch syndrome genes (MLH1, MSH2, MSH6 and PMS2) came next with 17 (5.3%) and were tied with TP53, also with 17 (5.3%). Next came ATM (15, 4.7%), CHEK2 (14, 4.4%), RAD51C (9, 2.8%), RAD51 (8, 2.5%), RAD50 (7, 2.2%), BRIP1 (6, 1.9%), BARD1 (4, 1.2%), PTEN (4, 1.2%), RAD51D (4, 1.2%) and APC (3, 0.9%). Other genes amounted 17 (3.0%) variants (NBN, RET, biallelic MUTYH, MITF, CDH1, EPCAM, FANCC, MRE11, POLD1, RB1, RECQL, MEN1, CDK4 and PTCH2). Moreover, monoallelic MUTYH variants, considered to have low penetrance, were detected in 42 (13.1%) cases (Fig. 1B).
In terms of unique variants, we detected 24 in BRCA1, 32 in BRCA2, 16 in PALB2, 16 in the Lynch syndrome genes, eight in TP53, 14 in ATM, eight in CHEK2, six in RAD51C, nine in RAD51, two in RAD50, six in BRIP1, four in PTEN, two in RAD51, three in APC, two in NBN, eight in MUTYH, two in CDH1 and one each in BARD1, RET, MITF, EPCAM, FANCC, MRE11, POLD1, RB1, RECQL, MEN1, CDK4, and PTCH2.
Among the 321 pathogenic/likely pathogenic variants, 84 (26.2%) were point mutations in conserved splice sites, 73 (22.7%) were frameshift deletions, 65 (20.2%) were missense point mutations, 58 (18.1%) were nonsense point mutations, 22 (6.9%) were frameshift duplications, 10 (3.1%) were frameshift insertions, four (1.3%) were frameshift indels, four inframe deletions (1.3%), and a single inframe insertion (0.3%) (Fig. 1C).
Most of the 1252 VUS (corresponding to 886 unique variants) were identified in ATM (198, 15.8%), followed by BRCA2 (86, 6.9%), MHS6 (63, 5.0%), BRIP1 (55, 4.4%), RAD50 (52, 4.2%), CHEK2 (38, 3.0%), PALB2 (34, 2.7%), MHS2 (31, 2.5%), MLH1 (17, 1.4%), TP53 (17, 1.4%), BRCA1 (17, 1.4%). Other genes amounted to 644 (51.4%) VUS (Fig. 1D).
Individuals with multiple variants (transheterozygotes)
Among the 305 individuals with pathogenic/likely pathogenic variants, 290 (95.1%) were single heterozygotes, 14 (4.6%) presented two variants in different genes (any two of these: APC, ATM, BRCA1, BRCA2, CDK4, CHEK2, MEN1, MUTYH, PALB2, PMS2, RAD51, RAD51C or TP53) and a single patient (0.3%) presented three pathogenic variants in different genes (BRCA1, MSH6, and MUTYH). The most common combination in the individuals with two variants was a variant in a high-penetrance gene, such as BRCA1 and BRCA2, alongside a variant with lower penetrance, such as monoallelic MUTYH or CHEK2. No individual presented more than one pathogenic/likely pathogenic variant in the same gene (Fig. 2, Supplementary Table 4). We observed a single individual BRCA1/BRCA2 double heterozygote, making the double heterozygotes prevalence among BRCA variants carriers about 0.78% (1/129) and 0.06% (1/1682) overall. Thus, our sample showed an overall transheterozygosity prevalence of about 0.89% (15/1682).

Heatmap representing the distribution of pathogenic/likely pathogenic (P/LP) transheterozygotes in the sample. The top line represents the number of patients in each column. The single heterozygotes (n = 290) are distributed in the main diagonal. Transheterozygotes (n = 15) are distributed in the inferior half (14 double-heterozygotes and 1 triple-heterozygote). The total number of variants are 321 (290 × 1 + 14 × 2 + 1 × 3 = 321).
Positivity profiles according to testing criteria of the USA National Comprehensive Cancer Network (NCCN) and Brazilian National Health Agency
We assessed the performance of the NCCN and Brazilian National Health Agency criteria for indication of genetic testing in our cohort. The sampled individuals were evaluated for hereditary cancer testing indication according to two sets of clinical criteria: the NCCN for hereditary breast/ovarian cancer syndromes, version 3.2019 and 1.2020 and the criteria of the Brazilian National Health Agency for breast and ovarian cancer syndrome (ANS 2018).
Among the 1682 individuals in this study, 306 did not have sufficient information for classification according to the guidelines and were excluded from this analysis. 1008 (59.9%) met NCCN 3.2019 and 1.2020 criteria for testing for HBOC syndrome genes, and 368 (21.9%) did not meet the criteria,. The positivity rate among individuals meeting NCCN criteria was 215/1008 (21.3%), while 45/360 (12.2%) of the individuals that did not meet NCCN criteria for testing were found to have P/LP variants. The true positive rate of NCCN criteria was 215/260 (82.7%) an the false negative rate was 45/260 (17.3%). The F1-measure was 33.9%.
Regarding ANS criteria, the 971 individuals with a personal history of breast or ovarian cancers (57.7%) met the criteria for testing, 418 (24.9%) did not meet and 293 (17.4%) did not have sufficient information for classification. The positivity rate for individuals meeting ANS criteria was 195/971 (20.1%), while 67/418 (16.0%) of the individuals that did not meet ANS criteria for testing were found to have P/LP variants. The true positive rate of ANS was 195/262 (77.4%) and the false negative rate was 67/262 (25.6%), and F1-measure was 31.6% (Table 2).
The group of 215 individuals fulfilling testing criteria according to the NCCN had the following genetic profiles: 72 (36.4%) individuals having variants in BRCA1, 29 (14.6%) in BRCA2, 16 (8.1%) in PALB2, 14 (7.1%) in TP53, 11 (5.6%) in ATM, 9 (4.5%) in CHEK2, 7 (3.5%) in RAD50 and RAD51, 6 (3.5%) in RAD51C, 4 (2.0%) in MSH6, 3 (1.5%) in BRIP1, MSH2 and RAD51D, 2 (1.0%) in APC, BARD1 and CDH1 e 8 (4.0%) in other genes (MEN1, MITF, MLH1, NBN, PMS2, POLD1, PTEN and RECQL).
The group of 45 individuals who did not meet the NCCN criteria had the following profiles: 9 (20.0%) individuals with variants in MUTYH, 7 (16.7%) in BRCA2, 6 (14.3%) in BRCA1, 5 (11.9%) in PALB2, 2 (4.8%) in ATM, CHEK2, MLH1, MSH6, PTEN, RET and TP53 each, besides 10 (23.8%) in other genes (BARD1, BRIP1, CDK4, FANCC, MRE11A, MSH2, PMS2, PTCH2, RAD51, and RAD51C).
In summary, both NCCN 1.2020/3.2019 and ANS 2018 criteria failed to detect a substantial part of positive individuals. The Brazilian criteria fared even worse, missing about 25% of positive individuals, versus approximately 17% with NCCN 1.2020/3.2019.
Geographic distribution of variants
We stratified our data according to each Brazilian state. The results are available in Supplementary Table 5.
Discussion
About 5–10% of cancer patients carry germline pathogenic variants in cancer predisposition genes. Identifying these patients is important because early diagnosis of hereditary cancer risk syndromes may improve vigilance and treatment. Therefore, the genetic investigation by NGS is already a tool of modern oncology [4].
Here, we report the results of our study, the largest conducted in Brazil, with 1682 individuals that underwent genotyping with 27- to 78-genes panels intended to detect germinative variants in cancer predisposition genes. Most patients in our study had a personal or family history of breast and ovarian cancer. Consequently, women are the majority in our sample (92.6%). Colorectal (4.2%), and thyroid/parathyroid (2.1%) were the second and third most prevalent cancers in this cohort.
The overall positivity rate in this study was 18.1% (305/1682 individuals). If we remove the individuals harboring the low penetrance monoallelic MUTYH variants, positivity drops to 16.0% (269/1682). Restricting the analysis to BRCA1/BRCA2 pathogenic/likely pathogenic variants only, the positivity rate was 7.7% (129/1682 individuals). This demonstrates the importance of comprehensive NGS multigene panels: it reduces the rate of false negatives, providing more information for oncologic management and prognosis.
The overall positivity rate of our study was slightly superior to other recently published data. Susswein et al. (2016) [5] found an overall yield of 9.0% among 10,046 patients referred for panel testing for hereditary cancers. Similarly, among 23,179 patients who received a 30-gene panel for hereditary cancer risk, 11.6% were found to have pathogenic variants [6]. In another study with 20,592 patients with breast cancer who had multigene panel testing, 10.2% were found to have pathogenic/ likely pathogenic variants. Our higher positivity rates may reflect that most individuals had a personal or family history of cancer.
BRCA1/2 positivity rate from our study was aligned with that seen in other studies. Indeed, since there is significant allelic heterogeneity in BRCA genes, there is great variability in positivity rates in other studies [7]. In Brazil, studies of HBOC patients observed positivity rates between 1.3% [8] and 27.3% [9]. In other countries, the positivity rate among sporadic and high-risk hereditary cancer cases varied between 2.6% in the USA [5] and 27.9% in Japan [10].
We compared the mutational profile observed in our study with that seen in other Brazilian studies. Nineteen variants in BRCA1, 14 in BRCA2, two in MUTYH, and one each in CHEK2, MSH2, MSH6, MLH1 and PALB2 were recurrent in other studies. These observations could mean that these are representatives from the mutational spectrum in Brazil and are important co-players in the risk of hereditary cancers in Brazilian populations (Supplementary Table 6).
The recurrence in independent samples from Brazil indicate the variants are representative of the BRCA1 and BRCA2 mutational spectrum in Brazil. Indeed, the BRCA1, c.5266dupC and BRCA2, c.156_157insAlu variants are considered as founder mutations coming from European populations and may be found in the individuals from all regions of Brazil, although at the same time there is also some heterogeneity between Brazilian regions, likelihood of finding certain variants in certain regions of the country [11]. Indeed, we detected BRCA1: c.5266dupC in individuals from Pernambuco and Alagoas, Northeast Brazil states, as well as in São Paulo (Southeast) and Paraná (South) and BRCA2:c.156_157insAlu was detected in an individual from Piauí, also a Northeast state.
Transheterozygosity, i.e. heterozygosity at two different loci [12], is rare among patients at risk of hereditary cancers. The prevalence of double heterozygosity among BRCA1/BRCA2 carriers ranges between 1.8% and 1.85% [13, 14] in the Jewish Ashkenazi populations. Among non-Ashkenazi Europeans, the prevalence is lower, between 0.22% and 0.87% [13]. An Italian study reported 0.62% [15] and a Korean study reported 1.2% [16]. Therefore, our estimate of 0.78% in the Brazilian sample seems plausible. BRCA1/BRCA2 double heterozygote may have early onset of disease and the phenotype is perhaps similar to the “severe end of spectrum of BRCA1 mutation carriership” [13]. We found a single BRCA1/BRCA2 double heterozygote woman in our sample. She presented personal history of breast cancer. Since she is in her late 40 s, her first cancer case must have happened much earlier, however, detailed data on her phenotype is currently lacking.
Literature on BRCA/other genes double heterozygotes is sparser. Thus, comparison with our results was difficult due to the possible different combinations of variants. For example, Sokolenko et al. [17] found seven digenic combinations double heterozygotes among BRCA1 and other DNA double-strand repair genes (BRCA1/CHEK2, BRCA1/ATM, BRCA1/BLM, CHEK2/BLM, CHEK2/ATM, NBS1/ATM, and NBS1/BLM) in Russian patients: none were observed in our study. They did not observe differences from single heterozygote individuals. In contrast, a case series including a German BRCA1/PALB2 double heterozygous patient had no early onset but had severe disease (multifocal triple negative ductal carcinoma) [18]. Another case report presented a case of a double heterozygote APC/MLH1 man with Kashmir/Egyptian ancestry, which had a history of six jejunal cancers [19]. Therefore, double heterozygosity may have unusual effects on cancer phenotypes.
We found that NCCN 1.2020 criteria missed a substantial proportion of individuals that had pathogenic/likely pathogenic variants. Other authors observed the same. We found that NCCN criteria missed a substantial proportion of individuals that had pathogenic/likely pathogenic variants. Other authors observed the same. For example, Grindedal et al. [20] investigated BRCA mutations in a Norwegian breast cancer cohort and assessed some testing criteria, including NCCN, and found a false negative rate of 15.8%. Yang et al. [21] investigated 4196 patients genotyped with 40- to 80- genes panels and showed a false negative rate of 13.5% with NCCN criteria. Both were not much far from our estimate of 17% with NCCN 1.2020/3.2019.
Other authors proposed changes to NCCN criteria. Alemar et al. [22] found that adding criteria that are not included in the NCCN and ANS criteria (e.g. some ASCO criteria [23, 24]) achieved a higher predictive value, while other authors compared other four algorithms (BOADICEA [25], BRCAPRO, Myriad [26] and Manchester score [27]) and observed that the pedigree-based BOADICEA most accurately predicted BRCA1/BRCA2 variant carrier status in a Southeastern Brazilian population [28]. Although NCCN criteria were imperfect, ANS criteria fared worse and a reformulation is warranted.
Recently, a panel of Brazilian experts proposed recommendations for improving testing criteria for HBOC risk patients in Brazil. Besides modifying testing criteria, the expert panel also recommended offering risk-reducing surgeries for positive patients. For negative patients, investigating both maternal and paternal lineages is warranted, so the result of models estimating cancer risk can be communicated to the patient. They also suggested that VUS should always be reported and periodically reassessed, but no urgent clinical action is justified since most VUS are constantly reclassified to benign/likely benign categories. Furthermore, patients should be contacted whenever any update in testing protocols or management options should appear [29].
Our study had some limitations. The NGS panel detects small deletions and duplications up to 17 base-pairs, but large deletions and duplications are not detected by this methodology. Other structural chromosomal changes, such as inversions and translocations are not detected either. If some of these changes are suspected, we recommend using methodologies like array CGH, MLPA, qPCR or FISH to confirm the variant found. Expansion variants of trinucleotide repeats, deep intronic variants or regulatory regions such as promoters are not detectable in the present test. Epigenetic changes are also not detectable by this test. At least for BRCA genes though, large rearrangements seem to be uncommon in Brazilian populations [11, 30]. Moreover, further phenotype information was missing for several patients, precluding further analyses. Since most individuals in our sample were female and had personal or familial history of breast or ovarian cancer, the results may be biased towards these types of cancers, but the data generated in this study is undoubtedly valuable for other types of cancer.
In conclusion, we genotyped 1682 Brazilian individuals referred for testing for hereditary cancer syndromes from all regions of the country with NGS multigenic panels suited for the detection of germline pathogenic variants associated with cancer susceptibility, making it the largest Brazilian study of this nature to date. We observed several BRCA1 and BRCA2 recurrent mutations, confirming their presence in the Brazilian mutational spectrum and generated data for other 30 genes and 110 variants with modest penetrance. We also estimated the prevalence of BRCA as well as non-BRCA double heterozygotes in the Brazilian population. More studies are necessary to discover the implication of transheterozygosity over the phenotype of affected individuals.
Data availability
All data generated or analysed during this study are included in this published article and its Supplementary Information Files.
References
Nagy R, Sweet K, Eng C. Highly penetrant hereditary cancer syndromes. Oncogene. 2004;23:6445–70.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Biesecker LG, Harrison SM. ClinGen Sequence Variant Interpretation Working G. The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet Med. 2018;20:1687–8.
Yoshida R. Hereditary breast and ovarian cancer (HBOC): review of its molecular characteristics, screening, treatment, and prognosis. Breast Cancer. 2020;28:1167–80.
Susswein LR, Marshall ML, Nusbaum R, Vogel Postula KJ, Weissman SM, Yackowski L, et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet Med. 2016;18:823–32.
Neben CL, Zimmer AD, Stedden W, van den Akker J, O’Connor R, Chan RC, et al. Multi-gene panel testing of 23,179 individuals for hereditary cancer risk identifies pathogenic variant carriers missed by current genetic testing guidelines. J Mol Diagn. 2019;21:646–57.
Alemar B, Herzog J, Brinckmann Oliveira Netto C, Artigalás O, Schwartz IVD, Matzenbacher Bittar C, et al. Prevalence of Hispanic BRCA1 and BRCA2 mutations among hereditary breast and ovarian cancer patients from Brazil reveals differences among Latin American populations. Cancer Genet. 2016;209:417–22.
Dillenburg CV, Bandeira IC, Tubino TV, Rossato LG, Dias ES, Bittelbrunn AC, et al. Prevalence of 185delAG and 5382insC mutations in BRCA1, and 6174delT in BRCA2 in women of Ashkenazi Jewish origin in southern Brazil. Genet Mol Biol. 2012;35:599–602.
Cipriano NM Jr., de Brito AM, de Oliveira ES, de Faria FC, Lemos S, Rodrigues AN, et al. Mutation screening of TP53, CHEK2 and BRCA genes in patients at high risk for hereditary breast and ovarian cancer (HBOC) in Brazil. Breast Cancer. 2019;26:397–405.
Okano M, Nomizu T, Tachibana K, Nagatsuka M, Matsuzaki M, Katagata N, et al. The relationship between BRCA-associated breast cancer and age factors: an analysis of the Japanese HBOC consortium database. J Hum Genet. 2021;66:307–14.
Palmero EI, Carraro DM, Alemar B, Moreira MAM, Ribeiro-Dos-Santos A, Abe-Sandes K, et al. The germline mutational landscape of BRCA1 and BRCA2 in Brazil. Sci Rep. 2018;8:9188.
Rebbeck TR, Friebel TM, Mitra N, Wan F, Chen S, Andrulis IL, et al. Inheritance of deleterious mutations at both BRCA1 and BRCA2 in an international sample of 32,295 women. Breast Cancer Research. 2016;18:112.
Leegte B, van der Hout AH, Deffenbaugh AM, Bakker MK, Mulder IM, ten Berge A, et al. Phenotypic expression of double heterozygosity for BRCA1 and BRCA2 germline mutations. J Med Genet. 2005;42:e20.
Lavie O, Narod S, Lejbkowicz F, Dishon S, Goldberg Y, Gemer O, et al. Double heterozygosity in the BRCA1 and BRCA2 genes in the Jewish population. Ann Oncol. 2011;22:964–6.
Zuradelli M, Peissel B, Manoukian S, Zaffaroni D, Barile M, Pensotti V, et al. Four new cases of double heterozygosity for BRCA1 and BRCA2 gene mutations: clinical, pathological, and family characteristics. Breast Cancer Res Treat. 2010;124:251–8.
Noh JM, Choi DH, Nam SJ, Lee JE, Kim JW, Kim SW, et al. Characteristics of double heterozygosity for BRCA1 and BRCA2 germline mutations in Korean breast cancer patients. Breast Cancer Res Treat. 2012;131:217–22.
Sokolenko AP, Bogdanova N, Kluzniak W, Preobrazhenskaya EV, Kuligina ES, Iyevleva AG, et al. Double heterozygotes among breast cancer patients analyzed for BRCA1, CHEK2, ATM, NBN/NBS1, and BLM germ-line mutations. Breast Cancer Res Treat. 2014;145:553–62.
Pern F, Bogdanova N, Schürmann P, Lin M, Ay A, Länger F, et al. Mutation analysis of BRCA1, BRCA2, PALB2 and BRD7 in a hospital-based series of German patients with triple-negative breast cancer. PLoS One. 2012;7:e47993.
Lindor NM, Smyrk TC, Buehler S, Gunawardena SR, Thomas BC, Limburg P, et al. Multiple jejunal cancers resulting from combination of germline APC and MLH1 mutations. Fam Cancer. 2012;11:667–9.
Grindedal EM, Heramb C, Karsrud I, Ariansen SL, Mæhle L, Undlien DE, et al. Current guidelines for BRCA testing of breast cancer patients are insufficient to detect all mutation carriers. BMC Cancer. 2017;17:438.
Yang S, Axilbund JE, O’Leary E, Michalski ST, Evans R, Lincoln SE, et al. Underdiagnosis of hereditary breast and ovarian cancer in medicare patients: genetic testing criteria miss the mark. Ann Surg Oncol. 2018;25:2925–31.
Alemar B, Gregório C, Herzog J, Matzenbacher Bittar C, Brinckmann Oliveira Netto C, Artigalas O, et al. BRCA1 and BRCA2 mutational profile and prevalence in hereditary breast and ovarian cancer (HBOC) probands from Southern Brazil: Are international testing criteria appropriate for this specific population? PLOS ONE. 2017;12:e0187630.
American Society of Clinical Oncology. Statement of the American Society of Clinical Oncology: genetic testing for cancer susceptibility, Adopted on February 20, 1996. J Clin Oncol. 1996;14:1730–6. discussion 7-40
Lu KH, Wood ME, Daniels M, Burke C, Ford J, Kauff ND, et al. American Society of Clinical Oncology Expert Statement: collection and use of a cancer family history for oncology providers. J Clin Oncol. 2014;32:833–40.
Antoniou AC, Pharoah PP, Smith P, Easton DF. The BOADICEA model of genetic susceptibility to breast and ovarian cancer. Br J Cancer. 2004;91:1580–90.
Parmigiani G, Berry D, Aguilar O. Determining carrier probabilities for breast cancer-susceptibility genes BRCA1 and BRCA2. Am J Hum Genet. 1998;62:145–58.
Evans DG, Eccles DM, Rahman N, Young K, Bulman M, Amir E, et al. A new scoring system for the chances of identifying a BRCA1/2 mutation outperforms existing models including BRCAPRO. J Med Genet. 2004;41:474–80.
Teixeira N, Maistro S, Del Pilar Estevez Diz M, Mourits MJ, Oosterwijk JC, Folgueira MAK, et al. Predictability of BRCA1/2 mutation status in patients with ovarian cancer: How to select women for genetic testing in middle-income countries. Maturitas. 2017;105:113–8.
Achatz MI, Caleffi M, Guindalini R, Marques RM, Nogueira-Rodrigues A, Ashton-Prolla P. Recommendations for advancing the diagnosis and management of hereditary breast and ovarian cancer in Brazil. JCO Glob Oncol. 2020;6:439–52.
Ewald IP, Cossio SL, Palmero EI, Pinheiro M, Nascimento IL, Machado TM, et al. BRCA1 and BRCA2 rearrangements in Brazilian individuals with hereditary breast and ovarian cancer syndrome. Genet Mol Biol. 2016;39:223–31.
Funding
No specific funding was received.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This project was approved by the Research Ethics Committee of the Institute of Integral Medicine Professor Fernando Figueira (CEP-IMIP) in Recife, Pernambuco and all individuals provided written consent for multi-gene testing (protocol number 29567220.4.1001.0071).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
de Oliveira, J.M., Zurro, N.B., Coelho, A.V.C. et al. The genetics of hereditary cancer risk syndromes in Brazil: a comprehensive analysis of 1682 patients. Eur J Hum Genet 30, 818–823 (2022). https://doi.org/10.1038/s41431-022-01098-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-022-01098-7
This article is cited by
-
Clinical genomics testing: mainstreaming and globalising
European Journal of Human Genetics (2022)
