
Abstract
The additional sex combs-like (ASXL) gene family—encoded by ASXL1, ASXL2, and ASXL3—is crucial for mammalian development. Pathogenic variants in the ASXL gene family are associated with three phenotypically distinct neurodevelopmental syndromes. Our previous work has shown that syndromic conditions caused by pathogenic variants in epigenetic regulatory genes show consistent patterns of genome-wide DNA methylation (DNAm) alterations, i.e., DNAm signatures in peripheral blood. Given the role of ASXL1 in chromatin modification, we hypothesized that pathogenic ASXL1 variants underlying Bohring-Opitz syndrome (BOS) have a unique DNAm signature. We profiled whole-blood DNAm for 17 ASXL1 variants, and 35 sex- and age-matched typically developing individuals, using Illumina’s Infinium EPIC array. We identified 763 differentially methylated CpG sites in individuals with BOS. Differentially methylated sites overlapped 323 unique genes, including HOXA5 and HOXB4, supporting the functional relevance of DNAm signatures. We used a machine-learning classification model based on the BOS DNAm signature to classify variants of uncertain significance in ASXL1, as well as pathogenic ASXL2 and ASXL3 variants. The DNAm profile of one individual with the ASXL2 variant was BOS-like, whereas the DNAm profiles of three individuals with ASXL3 variants were control-like. We also used Horvath’s epigenetic clock, which showed acceleration in DNAm age in individuals with pathogenic ASXL1 variants, and the individual with the pathogenic ASXL2 variant, but not in individuals with ASXL3 variants. These studies enhance our understanding of the epigenetic dysregulation underpinning ASXL gene family-associated syndromes.
Similar content being viewed by others

Introduction
The additional sex combs-like (ASXL) gene family includes ASXL1, ASXL2, and ASXL3 which encode epigenetic scaffolding proteins [1, 2]. Pathogenic variants in ASXL genes cause three distinct chromatinopathies that present clinically as neurodevelopmental disorders (NDDs) [2]. Bohring-Opitz syndrome (BOS; [MIM# 605039]) is caused by autosomal dominant truncating variants in ASXL1 and was molecularly defined in 2011 [3, 4]. BOS is characterized by poor growth, microcephaly, dysmorphic facial features, typical “BOS posture”, variable but typically severe to profound intellectual disability, feeding difficulties, and seizures [4, 5]. More recently, germline pathogenic variants in ASXL2 (reported in 2016) and ASXL3 (reported in 2013) were identified as the causes for Shashi-Pena syndrome (SHAPNS; MIM# 617190]) and Bainbridge-Ropers syndrome (BRS; MIM# 615485), respectively [6, 7].
Somatic truncating variants in ASXL1 are associated with age-related clonal hematopoiesis as well as various cancers [8]. Individuals with BOS have been reported to have Wilms tumor, suggesting that germline ASXL1 mutations increase the risk for certain cancers [1, 4]. The clinical interpretation of ASXL1 variants can be challenging, particularly when relying on population databases such as the genome aggregation database (gnomAD) [9]. Pathogenic variants in ASXL1 are reported at higher frequencies in the population than expected, partly due to acquired somatic mosaicism during hematopoietic clonal expansion which occurs in aging healthy individuals. Current tools for variant interpretation can introduce further ambiguity, emphasizing the need for robust and orthogonal tools for variant classification.
In 1998, studies in Drosophila demonstrated that the Asx gene family regulates transcription of developmentally vital genes such as the Hox gene cluster [1, 8]. Later studies showed the ASXL gene family has a more extensive role in epigenetic and transcriptional regulation [8]. In humans, ASXL1 is expressed in all tissues and interacts with the BRCA1-associated protein 1 (BAP1) to form the polycomb repressive deubiquitination complex (PR-DUB) [10]. The PR-DUB complex deubiquitinates histone 2A lysine 119 (H2AK119ub), a mark deposited by the Polycomb Repressive Complex (PRC1) [10]. Accumulating evidence also suggests a role for ASXL1 in regulating H3K27 trimethylation through interactions with components of the PRC2 [11]. More recent data show that ASXL1 interacts with all members of the cohesin complex required for chromatid separation and transcriptional regulation [12], further expanding the catalog of ASXL1 transcriptional and epigenetic co-regulators.
Our research group and others have shown that neurodevelopmental disorders caused by pathogenic variants in genes encoding epigenetic regulators can be associated with genome-wide changes in DNA methylation (DNAm), termed “DNAm signatures” [13,14,15,16,17,18,19]. To date, DNAm signatures for more than > 50 disorders have been defined. Most of these disorders are caused by variants in genes encoding histone modifying enzymes. These DNAm signatures are likely established via crosstalk between histone modifications and DNA methylation. Although the exact mechanisms underpinning DNAm signatures are not yet fully elucidated [20], a rapidly expanding body of work has emerged demonstrating that DNAm signatures have diagnostic utility in classifying variants of uncertain significance (VUS) [13,14,15,16,17,18,19].
For VUS classification, the DNAm profile for a single case is compared to a gene-specific DNAm signature. This single case analysis can be extended to compare DNAm profiles of individuals with sequence variants in functionally overlapping genes or other phenotypically overlapping neurodevelopmental disorders [13,14,15,16,17,18,19]. For example, the Weaver syndrome DNAm signature not only positively classifies individuals with pathogenic variants in EZH2 but also those with pathogenic variants in EED and SUZ12 [17]. These three genes encode components of the polycomb repressive complex 2 (PRC2) and are associated with clinically overlapping syndromes. In contrast to VUS classification, the derivation of a robust DNAm signature requires DNA from a cohort of individuals with a specific clinical diagnosis as well as pathogenic variants in the associated gene [21].
Here, we generated a unique DNAm signature for pathogenic ASXL1 variants in a cohort of individuals with BOS. We then used this signature to classify ASXL1 VUS and a small number of available pathogenic variants in ASXL2 and ASXL3. The ASXL2 variant had an overlapping DNAm profile with ASXL1 variants, defining significant congruence of epigenetic dysregulation for pathogenic variants in ASXL1 and ASXL2 but not ASXL3 variants. We also identified increased epigenetic age acceleration in individuals with pathogenic ASXL1 variants.
Methods
Research participants
Individuals were recruited through a patient registry at The University of California, Los Angeles USA in collaboration with the ARRE (ASXL Rare Research Endowment). Individuals with missense ASXL1 variants were identified through MSSNG [22], the largest whole genome sequencing [WGS] project for Autism Spectrum Disorder (ASD); SFARI [23] (Simons Foundation Autism Research Initiative) and the Simons Simplex Collection (SSC) using the Genotypes and Phenotypes in Families (GPF) tool (https://gpf.sfari.org/). We identified and recruited 17 individuals carrying ASXL1 variants, 1 individual carrying an ASXL2 variant, and 3 individuals carrying ASXL3 variants. Participant’s demographic, clinical phenotype, and variant information are in Table S1. We split individuals with classic features of BOS and pathogenic variants in ASXL1 (n = 14) into DNAm signature discovery n = 8 and validation n = 6 cohorts. In the ASXL1 cohort, three (n = 3) individuals carried missense VUS that were included for classification. We also included individuals with truncating variants in ASXL2 (n = 1) and ASXL3 (n = 3) for classification. Banked DNA samples from age- and sex-matched typically developing individuals (n = 135) were included as control subjects. These individuals were recruited from the Hospital for Sick Children and the Province of Ontario Neurodevelopmental Disorders (POND) Network and were deemed typically developing (Dr. Gregory Hanna). “Typically developing” was defined as healthy and developmentally normal by using formal cognitive/behavioral assessments (POND) or via physician/parental screening questionnaires (SickKids).
DNA methylation profiling and data processing
Genomic DNA was extracted from peripheral blood and bisulfite converted using the EpiTect Bisulfite Kit (EpiTect PLUS Bisulfite Kit, QIAGEN). Sodium bisulfite converted DNA was then hybridized to the Illumina Infinium Human Methylation EPIC BeadChip to interrogate more than 850,000 CpG sites in the human genome at The Center for Applied Genomics (TCAG), Hospital for Sick Children Research Institute. Samples were run in a single batch to reduce batch effects. On each microarray chip, cases and controls were randomly assigned a chip position. The minfi Bioconductor package in R was used to preprocess data including quality control, Illumina normalization and background subtraction, followed by extraction of beta (β) values [24]. Standard quality control metrics in minfi were used, including median intensity QC plots, density plots, and control probe plots; three ASXL1 samples (EX10, EX11, EX13) had lower median channel intensity values than recommended by minfi standards, and were used for signature validation but not discovery. Probes with detection flaws (n = 1061), probes near SNPs with minor allele frequencies above 1% (n = 29,958), cross-reactive probes (n = 41,975) [25], probes with raw beta of 0 or 1 in >0.25% of samples (n = 247), non-CpG probes (n = 2,925), and X and Y chromosome probes (n = 19,627) were removed, resulting in a total of n = 774,051 probes remained for differential methylation analysis.
DNA methylation age estimation
DNA methylation age (epigenetic age) was estimated using the calculator available for Illumina EPIC assays (http://dnamage.genetics.ucla.edu/) [26]. Estimates of age acceleration were calculated by subtracting the chronological age from the estimated DNAm age. We estimated DNAm age and calculated age acceleration in typically developing controls (n = 35), and individuals with variants in ASXL1 (n = 17), ASXL2 (n = 1) and ASXL3 (n = 3). We used a paired Wilcoxon’s test to assess mean differences between chronological and estimated DNAm age in each group. Then to assess whether mean estimates of age acceleration were significantly different between controls and individuals carrying variants in ASXL genes we used a Mann-Whitney U-Test.
DNA methylation signature derivation
To assess DNAm patterns, we identified differentially methylated sites in whole blood-derived DNA from n = 8 individuals carrying LOF variants in ASXL1 and a clinical diagnosis of BOS compared to 26 sex- and age-matched typically developing controls (Table S1 and S2). Sample numbers were not sufficient to generate robust signatures for ASXL2 or ASXL3 variants, we instead classified those samples using the generated BOS DNAm signature. For all samples, we applied the blood cell-type proportion estimation tool in minfi based on Illumina EPIC array data from FACS-sorted blood cells [26]. We identified differentially methylated CpG sites using Limma [27] regression modeling with age, sex, 5/6 cell type proportions (i.e., excluding neutrophils), and DNAm age residual (from Horvath DNAm age calculator) as covariates. The thresholds for differentially methylated CpG sites were Benjamini-Hochberg adjusted p value < 0.05 and a|Δβ| > 0.10. Δβ represents the difference in average DNAm (β) between groups. Principal component analysis (PCA) and hierarchical clustering were generated using Qlucore Omics Explorer (QOE, www.qlucore.com).
Machine learning classification models
We developed a machine learning model using the BOS DNAm signature. Using the R package ‘caret’, CpG sites with correlations equal to or greater than 90% to other signature CpGs were removed as previously described [14]. This led to a set of n = 546 non-redundant CpG sites. A support vector machine (SVM) model, trained on the non-redundant CpG sites, was set to “probability” mode to generate SVM scores ranging between 0 and 1 (0%–100%), classifying variants as “high” (score > 0.5) or “low” (score < 0.5). To test model specificity, EPIC array data from additional typically developing controls (n = 101) were scored. To test model sensitivity, we classified n = 6 validation samples, from individuals with a confirmed LOF variants in ASXL1 and a clinical BOS diagnosis. We also classified individuals with variants in ASXL2 (n = 1) and ASXL3 (n = 3) to further assess specificity of the model. Lastly, we classified individuals with Kabuki (n = 11), Sotos (n = 19) and Weaver (n = 30) syndromes, carrying pathogenic variants in KMT2D, NSD1, and EZH2, respectively.
Gene ontology analysis
The list of CpG positions comprising the DNAm signature was submitted to GREAT (Genomic Regions Enrichment of Annotations Tool) for gene ontology (GO) enrichment analysis [28]. Enrichment of each GO term within the gene list was calculated using a foreground/background hypergeometric test over genomic regions, using the set of CpG sites after minfi probe quality control (n = 774,051) as a background set. Overlapping genes were mapped using default GREAT settings with the following exceptions: the cut-off to annotate a CpG as overlapping with a gene (“distal gene mapping” setting) was set to 10 kb, and only enriched terms with three or more gene hits and FDR < 0.05 were reported. We predicted proteins bound to genomic loci using the ChIP-seq Atlas Enrichment Tool (https://chip-atlas.org/) [29].
Results
Molecular Genetics
In this study, we reported 14 individuals with ASXL1 (NM015338.5) variants that are predicted to adversely impact protein function and are classified as pathogenic using the ACMG variant classification guidelines [30] (Table 1 and S1). Variants reported are frameshift or nonsense located within the last two exons (11 and 12), the observed mutational hotspot in ASXL1. Variants in these individuals are associated with the phenotypes of Bohring-Opitz syndrome. The remaining variants (n = 3) in ASXL1 are missense variants of uncertain significance also located within the last two exons. Figure 1 provides a schematic ASXL1 structure and variant location generated using ProteinPaint [31].

Schematic representation of the ASXL1 protein (GenBank: ASXL1; NM_015338.6; GRCh37), its functional domains, and variants used in this study. Exon structure, based on GenBank: NM_015338.6, is provided by dashed lines. Red, HB1, ASXL, restriction endonuclease HTH domain (HARE-HTH, 11–83); purple, Asx homology domain (ASXH, 236–359); green, C-terminal plant homeodomain (PHD, 1506–1539). The N-terminal HARE-HTH domain is DNA binding and with the ASXH domain are required for interaction with BAP1 and NCOA1. The c-terminal PHD is required for interaction with nuclear receptors. The map was generated using ProteinPaint [31].
Epigenetic age in individuals with ASXL variants
Prior to generating a DNAm signature for ASXL1, we used the Horvath [26] DNAm age clock, to estimate epigenetic age for 35 typically developing controls and 21 individuals carrying variants in ASXL genes. Mean estimated DNAm age compared to mean chronological age was increased across all groups, however most significantly in individuals with ASXL1 variants (p-value = 3.8e−6) (Fig. 2A). We calculated DNAm (epigenetic) age acceleration by subtracting the chronological age from the estimated DNA methylation age. Mean epigenetic age acceleration was significantly increased in individuals carrying ASXL1 variants compared to typically developing controls (p-value = 1.1e−7; Mann-Whitney U-Test) (Fig. 2B). Two individuals carrying ASXL1 variants displayed the lowest age acceleration that also fell within the control range, and they were: subject EX17 (+1.11 years) with a missense variant p. (Ala627Val), and subject EX9 (+1.95 years) a 2-day old female with a frameshift variant p. (Gly696Argfs*11). The remaining two individuals with missense ASXL1 variants, EX15 and EX16, displayed increased epigenetic age acceleration (+6.5 and +5.9 years), but lower than the average age acceleration observed in individuals with pathogenic ASXL1 variants and BOS (+14.4 years). The individual with the ASXL2 variant displayed increased DNAm age and epigenetic age acceleration (+16.7 years) outside of the control range (Fig. 2). Lastly, the three individuals with ASXL3 variants showed no epigenetic age acceleration and were all within range of typically developing controls.

A Box plot comparing “DNA methylation age” (blue) derived from the Illumina 850 K data and reported chronological age (red), on the y-axis. Groups are indicated on the x-axis and include typically developing controls (n = 35), individuals with ASXL1 variants (n = 17), individual with ASXL2 variant (n = 1), and individuals with ASXL3 variants (n = 3). Each individual observation is plotted as a circle. To assess whether the mean difference between DNAm age and chronological age is statistically significant within each group we used a paired Wilcoxon test (*p-value < 0.05). (B) Box plot of epigenetic age acceleration (y-axis) obtained by subtracting the chronological age from the estimated DNAm age for each individual. To assess whether mean epigenetic age acceleration estimates are significantly different between controls and individuals carrying variants in ASXL genes we used a Mann-Whitney U-Test, except for ASXL2 with a n = 1.
Bohring-Opitz Syndrome (BOS) DNAm signature generation
To generate a BOS-specific DNAm signature, we profiled genome wide DNAm in blood from individuals with a confirmed BOS diagnosis due to pathogenic ASXL1 variants (n = 14; Table 1). The BOS discovery cohort (n = 8) included 4 females and 4 males with mean age at sample collection of 14.5 ± 10.5 years (range 4–31 years). The 26 sex- and age- matched control subjects included 13 females and 13 males and mean age at sample collection of 17.7 ± 9.4 years (range 4–35 years) (Table S2).
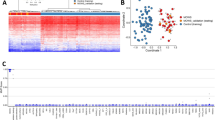
We identified 763 differentially methylated CpG sites that meet thresholds of FDR < 0.05 and|Δβ| > 0.10 (10% DNAm difference; Table S3), using linear regression modeling. We accounted for DNAm age as a covariate, considering the observed epigenetic age acceleration in individuals with ASXL1 variants. We visualized DNAm data at signature sites using principal component analysis (PCA) and hierarchal clustering (Fig. 3). DNAm at 763 signature sites clearly distinguished individuals with BOS from typically developing controls; 52% of the signature CpG sites were hypermethylated and 48% were hypomethylated. Approximately 55% of CpG signature sites overlapped islands or shores (within 2 kb of islands), this was significantly higher than the percentage of total probes on the array representing islands and shores (37%) (p-value = 9.68E−4; hypergeometric test).

A Principal component analysis (PCA) and (B) heatmap showing clustering of the BOS discovery cohort (n = 8; yellow) and control discovery cohort (n = 26; grey) using DNAm values at the 763 CpG sites identified in the BOS specific DNAm signature. The heatmap color gradient indicates the normalized DNAm value ranging from −2.0 (blue) to 2.0 (yellow). Euclidean distance metric is used in the heatmap clustering dendrograms.
Ontology of BOS DNAm signature sites
We assessed the ontology of genes overlapping CpG sites in the BOS DNAm signature using GREAT [28]. Using gene ontology analyses we can describe the role of gene targets in three biological domains: molecular-level activity of gene products (molecular functions), larger processes accomplished by multiple molecular functions (biological processes), and phenotypic abnormalities of human disorders that gene targets are predicted to contribute to (human phenotypes). There were 323 unique genes that overlapped 482 of the 763 signature CpG sites, with 86 genes overlapping 2 or more CpG sites. We identified significant enrichment (FDR < 0.05, gene hits ≥ 3) for 46 biological processes, 18 molecular functions, and 105 human phenotypes (Table S4–S6). The top 30 biological processes, ranked based on the number of gene hits, were related to embryonic bone, tissue, and organ development. Driving these biological processes were several HOX genes including HOXA5, HOXA11, and HOXB4. The HOXA5 gene was most notable, overlapping 13 CpG sites in the BOS DNAm signature, all of which were hypermethylated and mapped to a 3 kb window in CpG islands. In this study, we compared DNAm levels at eight of the 13 HOXA5 CpGs (within ~500 base pairs) in the BOS signature to DNAm values at those same sites in individuals with Kabuki, Weaver, and Sotos syndromes (Figure S1).
Most molecular functions were related to RNA polymerase II transcription factor activity and sequence-specific DNA-binding. The top 20 human phenotypes were related to bone abnormalities of the limbs and limb joints and included gene hits encoding collagens COL11A1 and COL6A1, homeobox proteins HOXA11, HOXA13 and MEIS1, and transcription factors RUNX2 and TWIST1. Lastly, we predicted proteins that bind the 763 BOS signature loci and the overlapping histone marks in blood using the ChIP-seq Atlas [29] enrichment tool. Top enriched peaks belonged to transcription factors: TET2, CTCF, the cohesin complex (RAD21, SMC3, SMC1A, and STAG1), and EZH2 from the PRC2 complex, and the top enriched histone mark was H3K27me3 (Table S7).
Independent validation of BOS DNAm signature
Using the BOS DNAm signature, we used a machine learning classification model to robustly categorize variants as BOS-like or control-like based on DNAm levels at signature sites. We trained a support vector machine (SVM) model on data from the discovery cohort used to generate the signature which included n = 8 individuals with BOS and typically developing controls (n = 26) (Table 1, S2). The model generated a probability of pathogenicity score from 0 to 1 for each sample, with 0.5 being the decision boundary for classification (Table S8). We classified a validation cohort of six unrelated individuals with a BOS diagnosis and truncating ASXL1 variants (Table 1). The SVM model generated high pathogenicity scores (87-98%) for the validation cohort demonstrating high sensitivity of the signature (Fig. 4). To test the specificity of the BOS DNAm signature, we included DNAm data for an additional 100 typically developing controls (40% females, and ages 1 to 42 years), all of which had low SVM scores (2–8%) demonstrating high specificity of the signature (Fig. 4).

Sample groups were scored using the BOS support vector machine (SVM) model. The x-axis groups each cohort, and the y-axis shows the probability score. BOS validation subject (n = 6) had high probability scores demonstrating 100% sensitivity of the model. Whereas validation control subjects (n = 101) all had low scores demonstrating 100% specificity of the model. ASXL1 missense variants (n = 3) and ASXL3 truncating variants (n = 3) scored low similar to controls, whereas the truncating ASXL2 variant (n = 1) scored high similar to the BOS validation group. Lastly, individuals with Sotos, Weaver, and Kabuki syndromes, caused by pathogenic variants in the chromatin-modifying genes NSD1, EZH2, and KMT2D respectively, all scored low further demonstrating 100% specificity of the model. Horizontal line represents threshold for classifying samples as case-like (above line) or control-like (below line).
Classification of variants in other ASXL genes causing neurodevelopmental disorders
The ASXL gene family also includes ASXL2 and ASXL3, and variants in these genes are implicated in Shashi-Pena and Bainbridge-Ropers syndromes, respectively [6, 7]. We used the SVM model to classify a truncating germline ASXL2 variant and three truncating germline ASXL3 variants (Table S1). The individual with the ASXL2 variant had a pathogenicity score of 84% on the SVM model and classified with the validation BOS cohort. In contrast, all individuals with truncating ASXL3 variants, had SVM scores of 6–7% within the range of controls (Table S8 and Fig. 4). We also visualized differences in DNAm for all individuals with ASXL variants based on the BOS DNAm signature sites using PCA and hierarchal clustering, and clustering patterns are in line with the SVM model classification (Figure S2).
To further validate the specificity of the BOS DNAm signature generated, we classified three cohorts of individuals with Sotos, Weaver, and Kabuki syndromes, caused by variants in genes encoding the epigenetic regulators, NSD1, EZH2, and KMT2D, respectively [13, 14, 17]. All three cohorts had pathogenicity scores within the control range (1–7%) (Fig. 4).
ASXL1 VUS Classification
Having illustrated the efficacy of the BOS DNAm signature in robustly classifying individuals with pathogenic ASXL1 variants, we next classified three individuals with de novo VUS in ASXL1. We also assessed predicted pathogenicity of these VUS using Alamut variant annotation software, which applies multiple prediction algorithms for comparison (Table 2). The SVM model for BOS DNAm signature sites generated low pathogenicity scores for all three missense variants (1-6%). For individual EX17 ASXL1 p. (Ala627Val), the DNAm data are in line with a non-BOS diagnosis. This individual is diagnosed with pervasive developmental disorders (PDD), attention deficit disorder (ADD), and Raynaud disease. This individual exhibits intellectual and speech delay but does not present with any of the hallmark features of BOS such as facial dysmorphism, growth restriction at birth, microcephaly, or recurrent infections. Clinical information available for individuals EX15 ASXL1 p. (Ser488Phe) and EX16 ASXL1 p. (Arg397His) was limited. Aside from a diagnosis of autism spectrum disorders (ASD), no other phenotypic information was available.
Discussion
In this study we profiled genome-wide DNA methylation in peripheral blood from 21 individuals with variants in ASXL genes. We used the DNAm data to (i) assess epigenetic age acceleration, (ii) generate a BOS-specific DNAm signature, and (iii) to classify ASXL1 VUS, and truncating ASXL2 and ASXL3 variants based on the unique BOS DNAm signature.
We report epigenetic age acceleration in individuals with BOS, but in not individuals with Bainbridge-Ropers syndrome; however, the latter could be due to a limited sample number (n = 3). Epigenetic age acceleration has previously been reported in Sotos syndrome and Tatton-Brown-Rahman syndrome (TBRS), which are caused by loss of function variants in two genes encoding epigenetic regulators, NSD1 and DNMT3A, respectively [32, 33]. NSD1 encodes a histone H3 lysine 36 (H3K36) methyltransferase responsible for regulating levels of trimethylation (H3K36me3), and DNMT3A encodes a de novo DNA methyltransferase [13, 33]. DNAm signatures for Sotos and TBRS indicate a primarily hypomethylated DNAm profile compared to typically developing controls with only a small proportion of hypermethylated sites [13, 33]. This aligns with global DNAm patterns observed during physiological aging, where the majority of the DNA is hypomethylated and preferentially hypermethylated at bivalent promoters [34]. This DNAm pattern is not consistent with the BOS DNAm signature we report, which has an approximately even number of hyper- and hypo- methylated CpG sites. While estimating epigenetic age can be an interesting biomarker, we don’t yet fully understand mechanisms driving DNAm changes and how they relate to physiological aging. However, estimating DNAm age in developmental disorders similar to BOS, will contribute to our understanding of DNAm changes related to aging.
Neurodevelopmental syndromes associated with ASXL genes are rare and have only recently been described. While the effects of ASXL genes variants have been studied in the context of somatic changes and cancer, the effects of germline ASXL variants in the context of the neurodevelopmental syndromes have not been well investigated. There are no iPSC models currently available for ASXL1 variants in BOS, and germline-modified mouse models have high rates of embryonic and postnatal mortality [35]. Most studies investigating effects of somatic ASXL1 variants are conducted in leukemic cell lines since these variants have been associated with acute myeloid leukemia (AML). Studies targeting ASXL1 gene expression in vitro have reported dysregulation of the posterior HOXA cluster (HOXA5, HOXA7, HOXA9) [11, 36, 37]. Several CpG sites in the BOS DNAm signature overlapped HOX genes, most notably 13 hypermethylated CpG sites in HOXA5, a homeobox sequence-specific transcription factor with finely tuned expression during development [38]. HOXA5 is a tumor suppressor gene dysregulated in expression and methylation in several types of cancer [39]. Our DNAm studies in Wilms tumor tissues identified hypermethylation of HOXA5 CpG sites [40, 41]. This finding is of interest since BOS individuals appear to have an increased risk for Wilms tumor [4]. In our previously published disorder-specific DNAm signatures we identified aberrant HOXA5 methylation in individuals with Weaver and Kabuki syndromes [14, 17]. In this study, we compared DNAm levels at HOXA5 CpGs in individuals with BOS, Kabuki, Weaver, and Sotos syndromes (Figure S1). Both individuals with BOS and Kabuki syndrome showed hypermethylation, whereas individuals with Weaver syndrome showed hypomethylation at these sites. Identifying recurrently affected HOX gene CpG sites across multiple NDDs defines critical regions that should be followed up by independent experimental approaches.
While research on the ASXL gene family has advanced rapidly, the exact mechanisms by which ASXL1 impacts DNA methylation of target genes have not been fully elucidated. In regards to the regulation of the posterior HOXA cluster by ASXL1, there is one proposed mechanism from a study in ASXL1-/- myeloid leukemia cells, which showed global depletion of H3K27me3 and EZH2 at the posterior HOXA cluster [11]. The same study reported direct physical interactions between ASXL1 and EZH2, a PRC2 protein responsible for depositing the H3K27me3 mark [11], and a protein that can interact with DNA methyltransferases thus impacting DNA methylation [42]. More recent ChIP-seq analyses of all epigenetic marks identified the co-occurrence of H3K27me3 at 74% of genomic regions with H2AK119ub marks [37]. Using the ChIP-seq Atlas enrichment tool [29], we predicted proteins bound to the 763 genomic loci in the signature and the overlapping histone marks. Interestingly the top enriched histone mark was H3K27me3 and EZH2 was one of the top enriched proteins bound (Table S7). Another potential mechanism by which ASXL1 can impact DNA methylation is through protein interactions with methyl-CpG binding domain proteins 5 and 6 (MBD5 and MBD6) [43]. MBD proteins coordinate crosstalk between DNA methylation, histone modifications, and chromatin organization [44]. These findings indicate that the actions of ASXL1 at a single genomic locus may result in a spectrum of epigenetic and transcriptional responses.
Previous DNAm signature studies in NDDs have contributed to our understanding of functional protein complexes and families. High or intermediate SVM classification of variants in one gene using the signature of another can occur when their protein products interact [1, 8, 21]. In addition to our findings in Weaver syndrome where variants in genes encoding components of PRC2 resulted in an overlapping DNAm profiles, individuals with variants in genes encoding components of the chromatin remodeling BAF complex, had overlapping DNAm profiles. Studies have shown that DNAm profiles at subsets of CpGs overlap between individuals with BAFopathies carrying variants in SMARCA2 that cause NCBRS, and in ARID1B, SMARCB1, and SMARCA4 that lead to Coffin-Siris syndromes type 1, 3, and 4, respectively. With respect to the ASXL gene family, phylogenetic analyses of coding sequences showed that ASXL1 and ASXL2 are more closely related than to ASXL3, and while ASXL1 and ASXL2 are expressed in most tissues, ASXL3 is strictly expressed in the brain. Studies have also shown co-immunoprecipitation of ASXL2 protein with BAP1, and that ASXL1 and ASXL2 can form mutually exclusive PR-DUB complexes with BAP1 [45, 46]. In this study, the DNAm profile of the individual with the truncating ASXL2 variant and a SHAPNS diagnosis had an overlapping DNAm profile to that of individuals with BOS. The individual with SHAPNS had a high pathogenicity SVM score (84%) based on the BOS DNAm signature and displayed epigenetic age acceleration, whereas the three individuals with truncating ASXL3 variants had low SVM scores (6–7%) and displayed no epigenetic age acceleration. While the overlapping DNAm signal is notable, we acknowledge that we have only one observation and the best practices for DNAm application include using only the gene-specific signature developed to classify VUS. However, ASXL2 germline variants are extremely rare, in part due to the recent identification of the SHAPNS clinical phenotypes, with only seven individuals reported in the literature [2, 7]. As more cases of ASXL2 are classified we expect to better define the congruence between the DNAm signatures for pathogenic variants in ASXL1 and ASXL2.
We used the BOS-specific DNAm signature to classify three individuals with ASXL1 VUS, all of which had pathogenicity SVM scores similar to those in controls. Clinical phenotype of individual EX17 p. (Ala627Val) is inconsistent with a BOS diagnosis, this is in line with the DNAm-negative classification for BOS (Table S1). To further test the specificity of the BOS DNAm signature we classified individuals with three syndromes: Sotos, Weaver, and Kabuki. Similar to ASXL1 in BOS, these syndromes are caused by variants in chromatin-modifying genes (NSD1, EZH2, KMT2D). Despite commonly dysregulated CpG sites across these syndromes, the BOS DNAm signature differentiated all individuals with these NDDs from those with BOS. This further demonstrates the specificity of the BOS DNAm signature.
In conclusion, we report an increased epigenetic age acceleration in individuals with BOS and a highly sensitive and specific BOS DNAm signature. These findings demonstrate the emerging trend in DNAm signature research, namely their utility beyond classifying VUS, in understanding and characterizing functional relationships between disorders. Future studies should include cell-type-specific multi-omics approaches which will be required to further elucidate molecular and epigenetic changes associated with ASXL1 variants causing BOS. Such studies will help determine whether there is a direct or indirect functional consequence of ASXL variants on DNAm and will also be useful in defining potential treatment targets for ASXL associated disorders.
Data availability
The datasets generated during the current study are not publicly available due to institutional ethical restrictions but are available from the corresponding author on reasonable request.
References
Katoh M. Functional and cancer genomics of ASXL family members. Br J Cancer. 2013;109:299–306.
Cuddapah VA, Dubbs HA, Adang L, Kugler SL, McCormick EM, Zolkipli-Cunningham Z, et al. Understanding the phenotypic spectrum of ASXL-related disease: Ten cases and a review of the literature. Am J Med Genet A. 2021;185:1700–11.
Hoischen A, van Bon BW, Rodríguez-Santiago B, Gilissen C, Vissers LE, de Vries P, et al. De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat Genet. 2011;43:729–31.
Russell B, Johnston JJ, Biesecker LG, Kramer N, Pickart A, Rhead W, et al. Clinical management of patients with ASXL1 mutations and Bohring-Opitz syndrome, emphasizing the need for Wilms tumor surveillance. Am J Med Genet A. 2015;167a:2122–31.
Russell BTW, Graham JM Jr. Bohring-Opitz Syndrome. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle; 1993–2021.
Bainbridge MN, Hu H, Muzny DM, Musante L, Lupski JR, Graham BH, et al. De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype with similarities to Bohring-Opitz syndrome. Genome Med. 2013;5:11.
Shashi V, Pena LD, Kim K, Burton B, Hempel M, Schoch K, et al. De Novo truncating variants in ASXL2 are associated with a unique and recognizable clinical phenotype. Am J Hum Genet. 2016;99:991–9.
Micol JB, Abdel-Wahab O. The role of additional sex combs-like proteins in cancer. Cold Spring Harb Perspect Med. 2016;6:a026526.
Carlston CM, O’Donnell-Luria AH, Underhill HR, Cummings BB, Weisburd B, Minikel EV, et al. Pathogenic ASXL1 somatic variants in reference databases complicate germline variant interpretation for Bohring-Opitz Syndrome. Hum Mutat. 2017;38:517–23.
Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 2010;465:243–7.
Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22:180–93.
Li Z, Zhang P, Yan A, Guo Z, Ban Y, Li J, et al. ASXL1 interacts with the cohesin complex to maintain chromatid separation and gene expression for normal hematopoiesis. Sci Adv. 2017;3:e1601602.
Choufani S, Cytrynbaum C, Chung BH, Turinsky AL, Grafodatskaya D, Chen YA, et al. NSD1 mutations generate a genome-wide DNA methylation signature. Nat Commun. 2015;6:10207.
Butcher DT, Cytrynbaum C, Turinsky AL, Siu MT, Inbar-Feigenberg M, Mendoza-Londono R, et al. CHARGE and Kabuki Syndromes: gene-specific DNA methylation signatures identify epigenetic mechanisms linking these clinically overlapping conditions. Am J Hum Genet. 2017;100:773–88.
Siu MT, Butcher DT, Turinsky AL, Cytrynbaum C, Stavropoulos DJ, Walker S, et al. Functional DNA methylation signatures for autism spectrum disorder genomic risk loci: 16p11.2 deletions and CHD8 variants. Clin Epigenetics. 2019;11:103.
Chater-Diehl E, Ejaz R, Cytrynbaum C, Siu MT, Turinsky A, Choufani S, et al. New insights into DNA methylation signatures: SMARCA2 variants in Nicolaides-Baraitser syndrome. BMC Med Genomics. 2019;12:105.
Choufani S, Gibson WT, Turinsky AL, Chung BHY, Wang T, Garg K, et al. DNA methylation signature for EZH2 functionally classifies sequence variants in three PRC2 complex genes. Am J Hum Genet. 2020;106:596–610.
Goodman SJ, Cytrynbaum C, Chung BH-Y, Chater-Diehl E, Aziz C, Turinsky AL, et al. EHMT1 pathogenic variants and 9q34.3 microdeletions share altered DNA methylation patterns in patients with Kleefstra syndrome. J Transl Genet Genomics. 2020;4:144–58.
Rots D, Chater-Diehl E, Dingemans AJM, Goodman SJ, Siu MT, Cytrynbaum C, et al. Truncating SRCAP variants outside the Floating-Harbor syndrome locus cause a distinct neurodevelopmental disorder with a specific DNA methylation signature. Am J Hum Genet. 2021;108:1053–68.
Janssen SM, Lorincz MC. Interplay between chromatin marks in development and disease. Nat Rev Genet. 2021;23:137–153.
Chater-Diehl E, Goodman SJ, Cytrynbaum C, Turinsky AL, Choufani S, Weksberg R. Anatomy of DNA methylation signatures: Emerging insights and applications. Am J Hum Genet. 2021;108:1359–66.
RK CY, Merico D, Bookman M, J LH, Thiruvahindrapuram B, Patel RV, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. 2017;20:602–11.
Fischbach GD, Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron 2010;68:192–5.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014;30:1363–9.
Chen YA, Choufani S, Grafodatskaya D, Butcher DT, Ferreira JC, Weksberg R. Cross-reactive DNA microarray probes lead to false discovery of autosomal sex-associated DNA methylation. Am J Hum Genet. 2012;91:762–4.
Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501.
Oki S, Ohta T, Shioi G, Hatanaka H, Ogasawara O, Okuda Y, et al. ChIP-Atlas: a data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 2018;19:e46255.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Zhou X, Edmonson MN, Wilkinson MR, Patel A, Wu G, Liu Y, et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet. 2016;48:4–6.
Martin-Herranz DE, Aref-Eshghi E, Bonder MJ, Stubbs TM, Choufani S, Weksberg R, et al. Screening for genes that accelerate the epigenetic aging clock in humans reveals a role for the H3K36 methyltransferase NSD1. Genome Biol. 2019;20:146.
Jeffries AR, Maroofian R, Salter CG, Chioza BA, Cross HE, Patton MA, et al. Growth disrupting mutations in epigenetic regulatory molecules are associated with abnormalities of epigenetic aging. Genome Res. 2019;29:1057–66.
Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–9.
Fisher CL, Lee I, Bloyer S, Bozza S, Chevalier J, Dahl A, et al. Additional sex combs-like 1 belongs to the enhancer of trithorax and polycomb group and genetically interacts with Cbx2 in mice. Dev Biol. 2010;337:9–15.
Asada S, Goyama S, Inoue D, Shikata S, Takeda R, Fukushima T, et al. Mutant ASXL1 cooperates with BAP1 to promote myeloid leukaemogenesis. Nat Commun. 2018;9:2733.
Balasubramani A, Larjo A, Bassein JA, Chang X, Hastie RB, Togher SM, et al. Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat Commun. 2015;6:7307.
Jeannotte L, Gotti F, Landry-Truchon K. Hoxa5: a key player in development and disease. J Dev Biol. 2016;4:13–31.
Bhatlekar S, Fields JZ, Boman BM. HOX genes and their role in the development of human cancers. J Mol Med. 2014;92:811–23.
Brzezinski J, Choufani S, Romao R, Shuman C, Chen H, Cunanan J, et al. Clinically and biologically relevant subgroups of Wilms tumour defined by genomic and epigenomic analyses. Br J Cancer. 2021;124:437–46.
Guerra J, Pereira BMS, Cruz J, Scherer NM, Furtado C, Montalvão de Azevedo R, et al. Genes controlled by DNA methylation are involved in Wilms tumor Progression. Cells. 2019;8:921-936.
Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006;439:871–4.
Baymaz HI, Fournier A, Laget S, Ji Z, Jansen PW, Smits AH, et al. MBD5 and MBD6 interact with the human PR-DUB complex through their methyl-CpG-binding domain. Proteomics 2014;14:2179–89.
Du Q, Luu PL, Stirzaker C, Clark SJ. Methyl-CpG-binding domain proteins: readers of the epigenome. Epigenomics 2015;7:1051–73.
Campagne A, Lee MK, Zielinski D, Michaud A, Le Corre S, Dingli F, et al. BAP1 complex promotes transcription by opposing PRC1-mediated H2A ubiquitylation. Nat Commun. 2019;10:348.
Sahtoe DD, van Dijk WJ, Ekkebus R, Ovaa H, Sixma TK. BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nat Commun. 2016;7:10292.
Acknowledgements
We are grateful to all the study participants and their families and the many clinicians who recruited them into this study. This work was supported by the ASXL Rare Research Endowment (ARRE) (MC2015-16), the Canadian Institutes of Health Research (CIHR) grants (IGH-155182 and MOP-126054), and the Ontario Brain Institute (Province of Ontario Neurodevelopmental Disorders (POND) network (IDS11-02)) grants to RW. The work was also supported by The Bohring-Opitz Syndrome Foundation and the ARRE (20201705) to VA and DP5OD024579 (NIH) to VA. We acknowledge the administrative assistance of Khadine Wiltshire, and the technical assistance of Youliang Lou, Chunhua Zhao, and Laila Sathe. Thank you as well to Dr. Greg Hanna for contributing blood DNA samples from typically developing control individuals who had undergone cognitive/behavioral assessments. The authors also wish to acknowledge the resources of MSSNG (www.mss.ng), Autism Speaks and The Centre for Applied Genomics at The Hospital for Sick Children, Toronto, Canada. We thank the participating families for their time and contributions to this database, as well as the generosity of the donors who supported this program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Informed consent was obtained from all research participants and/or their guardian(s) according to the protocol approved by the Research Ethics Board of the Hospital for Sick Children (REB# 1000038847) and UCLA (IRB #11-001087).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Awamleh, Z., Chater-Diehl, E., Choufani, S. et al. DNA methylation signature associated with Bohring-Opitz syndrome: a new tool for functional classification of variants in ASXL genes. Eur J Hum Genet 30, 695–702 (2022). https://doi.org/10.1038/s41431-022-01083-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-022-01083-0
This article is cited by
-
Chromatinopathies: insight in clinical aspects and underlying epigenetic changes
Journal of Applied Genetics (2024)
-
DNA methylation signatures for chromatinopathies: current challenges and future applications
Human Genetics (2024)
-
The omics era: a nexus of untapped potential for Mendelian chromatinopathies
Human Genetics (2024)
-
A new blood DNA methylation signature for Koolen-de Vries syndrome: Classification of missense KANSL1 variants and comparison to fibroblast cells
European Journal of Human Genetics (2024)
-
2022: the year that was in the European Journal of Human Genetics
European Journal of Human Genetics (2023)

{kind=link}