
Abstract
Isolated mitochondrial complex II deficiency is a rare cause of mitochondrial respiratory chain disease. To date biallelic variants in three genes encoding mitochondrial complex II molecular components have been unequivocally associated with mitochondrial disease (SDHA/SDHB/SDHAF1). Additionally, variants in one further complex II component (SDHD) have been identified as a candidate cause of isolated mitochondrial complex II deficiency in just two unrelated affected individuals with clinical features consistent with mitochondrial disease, including progressive encephalomyopathy and lethal infantile cardiomyopathy. We present clinical and genomic investigations in four individuals from an extended Palestinian family with clinical features consistent with an autosomal recessive mitochondrial complex II deficiency, in which our genomic studies identified a homozygous NM_003002.3:c.[205 G > A];[205 G > A];p.[(Glu69Lys)];[(Glu69Lys)] SDHD variant as the likely cause. Reviewing previously published cases, these findings consolidate disruption of SDHD function as a cause of mitochondrial complex II deficiency and further define the phenotypic spectrum associated with SDHD gene variants.
Similar content being viewed by others


Introduction
The mitochondrial oxidative phosphorylation (OXPHOS) system is composed of five multi-subunit transmembrane protein complexes (I–V) encoded by the mitochondrial and nuclear genomes, and is the primary mechanism for adenosine triphosphate production in eukaryotic cells. OXPHOS defects result in mitochondrial disease, with an estimated prevalence of 1:4300 [1, 2].
Mitochondrial complex II (succinate dehydrogenase) is composed of two catalytic subunits (SDHA/SDHB) anchored to the inner mitochondrial membrane by two smaller subunits (SDHC/SDHD) [3, 4]. Complex II differs from other mitochondrial respiratory chain complexes, in that the four structural subunits and their two assembly factors (SDHAF1/SDHAF2) are solely encoded by the nuclear genome. Complex II is also unique in being involved in both the mitochondrial respiratory chain and the Krebs cycle [2].
Mitochondrial complex II deficiency with multisystem involvement has been reported in association with biallelic SDHA [5], SDHB [2], SDHD [6, 7] and SDHAF1 [3, 8] gene variants, with clinical presentations including Leigh syndrome, leukoencephalopathy, optic atrophy and cardiomyopathy with highly variable severity and age of onset [5, 9]. Complex II deficiency is rare accounting for only 2–4% of OXPHOS defects [6], with variants in SDHA being most common, predominantly associated with Leigh syndrome [5]. Previously, only two individuals with candidate biallelic SDHD variants and isolated complex II deficiency have been reported [6, 7]. Here we describe four Palestinian siblings presenting in childhood with clinical features indicative of mitochondrial disease and a likely pathogenic homozygous SDHD variant, consolidating SDHD gene variants as a likely cause of autosomal recessive mitochondrial complex II deficiency.
Materials and methods
Blood samples were collected with informed consent (Palestinian Health Research Council; PHRC/HC/518/19). Single-nucleotide polymorphism (SNP) genotyping was performed (HumanCytoSNP-12 v2.1 Beadchip array: Illumina). Whole-exome sequencing (WES) (NextSeq1500: Illumina) analysis involved: Agilent SureSelect Whole Exome v6 targeting, read alignment (BWA-MEM,v0.7.17), mate-pairs fixed and duplicates removed (Picard v2.15.0), InDel realignment and base quality recalibration (GATK v3.7.0), SNVs/InDels (GATK/HaplotypeCaller), annotation using Alamut Batch (v1.10) and CNV detection with ExomeDepth [10] and Savvy CNV [11]. Dideoxy sequencing was undertaken using standard techniques.
Model 3abv (porcine heart mitochondrial complex II) [12] was selected from X-ray diffraction/NMR-derived structures of SDHD (O14521) and its homologues (RCSB Protein Data Bank). Amino acid residues were visualised with their polar bonds, and annotated using Pymol 2.3 [13, 14].
Results
Clinical findings
We describe four affected Palestinian patients (three male, one female) aged 4–20 years, comprising of two sibships from an extended interconnecting family (Fig. 1A). All four children presented with developmental delay in infancy and variable clinical and laboratory findings suggestive of a mitochondrial disorder including elevated serum lactate/urinary Krebs cycle metabolites, nystagmus, optic atrophy, progressive microcephaly, generalised hypotonia, epileptic seizures, severe/profound intellectual disability/developmental impairment and cardiomyopathy. The affected children were not dysmorphic (Fig. 1C), though individuals V:2 and V:4 were noted to have significant hypertrichosis, particularly over their back and limbs. MRI neuroimaging was unremarkable for one child at 8 months (V:2; Fig. 1D), however, his sister’s scan revealed delayed myelination at age 6 months (V:4). Hirschsprung disease, confirmed by aganglionic rectal biopsy, was noted in a single individual (V:2). A full description of the clinical features and disease progression is summarised in Table 1.

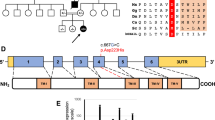
A Pedigree diagram showing segregation of the SDHD c.205 G > A; p.(Glu69Lys) variant. Genotypes are shown beneath generations IV and V (+, c.205 G > A; −, WT). Affected individuals were homozygous for SDHD c.205 G > A, DNA was available from all but one affected individual (V:5). B Electropherogram showing the DNA sequence at the position of SDHD c.205 G > A in a homozygous affected individual. C T1- and T2-weighted axial views of MRI head of individual V:2 (aged 8 months). Normal myelination and no intracranial abnormalities. D Image of affected individual V:4, illustrating the absence of any facial dysmorphism. E (i) Schematic localisation of SDHD p.(Glu69Lys), p.(Asp92Gly) and p.(Ter160LeuextTer3) variants within the succinate dehydrogenase cytochrome b small subunit (CybS) domain of the SDHD polypeptide. The orange rectangle denotes the transit peptide (TP) domain, the red circle denotes the iron (haem axial ligand) binding site shared with SDHC and the blue circle denotes the ubiquinone binding site shared with SDHB. (ii) Conservation of the SDHD p.(Glu69Lys), p.(Asp92Gly) and p.(Ter160LeuextTer3) variants across species. F Visual depiction of the two autozygous regions on chromosome 11 (shown in red) common to affected individuals V:2, V:4 and V:6 including the 2.37 Mb region containing 21 genes including SDHD.
Genetic findings and homology modelling
Genome-wide SNP genotyping and WES were undertaken assuming that a homozygous founder variant was responsible, although also considering other genetic mechanisms. SNP genotyping (individuals V:2, V:4 and V:6) identified four notable (>1 Mb) shared homozygous regions, the two largest identified on chromosome 11; a ~7.00 Mb region (rs6485795–rs11246414, Chr11:g.47908294–54905443 [hg38]) and a ~2.42 Mb region (rs120436–rs12794326, Chr11:g.110826521–113248134) (Fig. 1F). WES was performed on DNA from affected individual V:4, to identify rare functional candidate variants. Variants were prioritised by call quality and frequency (gnomAD v2.1.1/1000 Genomes Project, MAF ≤ 0.0001) and cross referenced with SNP data, identifying only a single candidate homozygous variant of relevance to the phenotype in SDHD NM_003002.3:c.[205G > A];[205G > A];p.[(Glu69Lys)];[(Glu69Lys)];Chr11:g.[112088902G > A];[112088902G > A], located within the second largest shared homozygous region. The variant is present in only two heterozygotes in gnomAD (v2.1.1) and is predicted to result in a glutamic acid—lysine substitution in an evolutionarily conserved Glu69 residue (Fig. 1E). This variant was previously reported as the likely candidate cause of disease in compound heterozygous form ((c.[205G > A];[479G > T]; p.[(Glu69Lys)];[(Ter160LeuextTer3)])) in a single individual with autosomal recessive encephalomyopathy and isolated mitochondrial complex II deficiency [6] (ClinVar accession:VCV000156153.8 and SCV001424558). Dideoxy sequencing confirmed cosegregation as appropriate for an autosomal recessive disorder (Fig. 1A, B ). Protein modelling positions the p.(Glu69Lys) substitution within the first transmembrane alpha helix, where it likely disrupts tertiary structure through interrupting a predicted hydrogen bond with Gln109 on the adjacent helix (Supplementary Fig. S1A). Conversely, a previously described variant, p.(Asp92Gly) is located at the apex of transmembrane alpha helices one and two, in close proximity to the membrane and inter-membrane space (Supplementary Fig. S1A, B).
Discussion
Here we define a homozygous SDHD c.[205G > A];[205G > A];p.[(Glu69Lys)];[(Glu69Lys)] missense variant as the likely cause of isolated mitochondrial complex II deficiency in three affected children from an extended Palestinian family. DNA was unavailable for V:5 (deceased age 10 years), whose clinical history overlapped that of his sibling (V:6). Tissues and organs heavily dependent on robust OXPHOS processes tend to be most affected by mitochondrial disease [15], explaining why common findings include optic atrophy, leukoencephalopathy, myopathy, cardiomyopathy and Leigh syndrome. These clinical features overlap those described in the two individuals with SDHD-related mitochondrial disease reported to date (Table 1). Previously, compound heterozygous variants in SDHD [6], including the same p.(Glu69Lys) variant identified here and a c.479G > T; p.(Ter160LeuextTer3) alteration (c.[205G > A];[479G > T];p.[(Glu69Lys)];[(Ter160LeuextTer3)]), were identified as the likely candidate cause of disease in a Swiss child presenting with developmental regression following a viral infection, at 3 months. Progressive ocular (visual impairment, nystagmus, optic disc pallor) and neurological (epileptic seizures, ataxia, dystonia and continuous intractable myoclonic movement) involvement were described, and the child died aged 10 years. Urinalysis revealed lactic aciduria, ketonuria and Krebs cycle intermediates. Complex II activity was deficient in skeletal muscle and complementation studies in patient fibroblasts showed restoration of complex II assembly and function with expression of wildtype, but not mutant, SDHD cDNA [6]. Subsequently an Irish male infant was described [7] homozygous for a novel SDHD c.[275A > G];[275A > G];p.[(Asp92Gly)];[(Asp92Gly)] substitution, presenting with cardiomyopathy in utero. He developed cardiopulmonary insufficiency rapidly after birth, dying on day 1 of life. Subsequent analysis of respiratory chain function in patient muscle homogenate revealed a marked defect in complex II activity.
The four affected individuals described here show phenotypic overlap with both these individuals (Table 1). Our study extends the clinical spectrum and highlights the wide range of phenotypical features and severity across affected individuals, even those with the same SDHD genotype (Table 1). Hypertrichosis, a recognised feature of some forms of mitochondrial disease (most notably SURF1-Leigh syndrome [16]), was a noted feature in two Palestinian children. Hirschsprung disease diagnosed in a single affected individual (V:2) has not been previously reported in association with SDHD variants, and it remains unclear whether this is an associated or unrelated feature. Neurodevelopmental regression is a common characteristic of mitochondrial disease, particularly during physiologic stress through intercurrent infection, prolonged fasting or dehydration [17]. It is thus unsurprising that this appears to be a common feature of complex II deficiency due to biallelic SDHD variants (Table 1). An accurate molecular diagnosis for complex II deficient patients would support avoidance of prolonged fasting and dehydration.
Homology modelling of the two putative pathogenic SDHD missense variants thus far associated with isolated autosomal recessive mitochondrial complex II deficiency [c.[205G > A];p.(Glu69Lys) and c.[275A > G];p.(Asp92Gly)] predicts disruption of non-covalent bonds between transmembrane helices and changes to complex II positioning in the inner mitochondrial membrane as likely outcomes (Supplementary Fig. S1a). In addition to their role in primary mitochondrial disease, heterozygous germline variants in other complex II subunits and assembly factors (including SDHA, SDHB, SDHC, SDHD and SDHAF2) are associated with paragangliomas, phaeochromocytomas and gastrointestinal stromal tumours [3]. None of the three SDHD variants associated with mitochondrial complex II deficiency have been previously linked to tumourigenesis, including in this extended Palestinian family, although a Dutch founder familial paraganglioma SDHD variant c.274G > T; p.(Asp92Tyr) has been described [18]. Additionally, SDHA and SDHB variants have been associated with both mitochondrial complex II deficiency in biallelic form, and hereditary cancer susceptibility in monoallelic form [5, 19]. Therefore routine surveillance of heterozygous SDHD carriers is suggested for early detection of paragangliomas and phaeochromocytomas and appropriate intervention. Together the data presented here consolidate biallelic SDHD variants as a cause of mitochondrial disease due to mitochondrial complex II malfunction, and extend the variable associated clinical features.
References
Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, Alston CL, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. 2015;77:753–9.
Alston CL, Davison JE, Meloni F, van der Westhuizen FH, He L, Hornig-Do HT, et al. Recessive germline SDHA and SDHB mutations causing leukodystrophy and isolated mitochondrial complex II deficiency. J Med Genet. 2012;49:569–77.
Hoekstra AS, Bayley JP. The role of complex II in disease. Biochim Biophys Acta. 2013;1827:543–51.
van den Heuvel L, Smeitink J. The oxidative phosphorylation (OXPHOS) system: nuclear genes and human genetic diseases. Bioessays. 2001;23:518–25.
Renkema GH, Wortmann SB, Smeets RJ, Venselaar H, Antoine M, Visser G, et al. SDHA mutations causing a multisystem mitochondrial disease: novel mutations and genetic overlap with hereditary tumors. Eur J Hum Genet. 2015;23:202–9.
Jackson CB, Nuoffer JM, Hahn D, Prokisch H, Haberberger B, Gautschi M, et al. Mutations in SDHD lead to autosomal recessive encephalomyopathy and isolated mitochondrial complex II deficiency. J Med Genet. 2014;51:170–5.
Alston CL, Ceccatelli Berti C, Blakely EL, Olahova M, He L, McMahon CJ, et al. A recessive homozygous p.Asp92Gly SDHD mutation causes prenatal cardiomyopathy and a severe mitochondrial complex II deficiency. Hum Genet. 2015;134:869–79.
Ghezzi D, Goffrini P, Uziel G, Horvath R, Klopstock T, Lochmuller H, et al. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat Genet. 2009;41:654–6.
Jain-Ghai S, Cameron JM, Al Maawali A, Blaser S, MacKay N, Robinson B, et al. Complex II deficiency-a case report and review of the literature. Am J Med Genet A. 2013;161A:285–94.
Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28:2747–54.
Laver TW, De Franco E, Johnson MB, Patel K, Ellard S, Weedon MN, et al. SavvyCNV: genome-wide CNV calling from off-target reads. bioRxiv. 2019:617605. https://doi.org/10.1101/617605.
Inaoka DK, Shiba T, Sato D, Balogun EO, Sasaki T, Nagahama M, et al. Structural insights into the molecular design of flutolanil derivatives targeted for fumarate respiration of parasite mitochondria. Int J Mol Sci. 2015;16:15287–308.
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–303.
Schrodinger L. The PyMOL molecular graphics system. 2010. Version. Version 2.3 Schrödinger, LLC. 2019.
Chinnery PF, Schon EA. Mitochondria. J Neurol Neurosurg Psychiatry. 2003;74:1188–99.
Ostergaard E, Bradinova I, Ravn SH, Hansen FJ, Simeonov E, Christensen E, et al. Hypertrichosis in patients with SURF1 mutations. Am J Med Genet A. 2005;138:384–8.
Falk MJ. Neurodevelopmental manifestations of mitochondrial disease. J Dev Behav Pediatr. 2010;31:610–21.
Hensen EF, van Duinen N, Jansen JC, Corssmit EP, Tops CM, Romijn JA, et al. High prevalence of founder mutations of the succinate dehydrogenase genes in the Netherlands. Clin Genet. 2012;81:284–8.
Grønborg S, Darin N, Miranda MJ, Damgaard B, Cayuela JA, Oldfors A, et al. Leukoencephalopathy due to complex II deficiency and Bi-Allelic SDHB mutations: further cases and implications for genetic counselling. JIMD Rep. 2016;33:69–77.
Acknowledgements
First and foremost, the authors are grateful to the Palestinian families for taking part in this study.
Funding
This work was supported by the University of Exeter Vice Chancellor Scholarship (SL), Wellcome Trust GW4-CAT Ph.D. Fellowship 220600/Z/20/z (JF), MRC Confidence in Concept MC_PC_18047 (ELB and AHC) and the Zamalah-Taawon Program Scholarship (RK-N).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, S., Fasham, J., Al-Hijawi, F. et al. Consolidating biallelic SDHD variants as a cause of mitochondrial complex II deficiency. Eur J Hum Genet 29, 1570–1576 (2021). https://doi.org/10.1038/s41431-021-00887-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00887-w
