
Abstract
Gastrointestinal stromal tumour (GIST) is a mesenchymal neoplasm arising in the gastrointestinal tract. A rare subset of GISTs are classified as wild-type GIST (wtGIST) and these are frequently associated with germline variants that affect the function of cancer predisposition genes such as the succinate dehydrogenase subunit genes (SDHA, SDHB, SDHC, SDHD) or NF1. However, despite this high heritability, familial clustering of wtGIST is extremely rare. Here, we report a mother–son diad who developed wtGIST at age 66 and 34 years, respectively. Comprehensive genetic testing revealed germline truncating variants in both SDHA (c.1534C>T (p.Arg512*)) and PALB2 (c.3113G>A (p.Trp1038*)) in both affected individuals. The mother also developed breast ductal carcinoma in-situ at age 70 years. Immunohistochemistry and molecular analysis of the wtGISTs revealed loss of SDHB expression and loss of the wild-type SDHA allele in tumour material. No allele loss was detected at PALB2 suggesting that wtGIST tumourigenesis was principally driven by succinate dehydrogenase deficiency. However, we speculate that the presence of multilocus inherited neoplasia alleles syndrome (MINAS) in this family might have contributed to the highly unusual occurrence of familial wtGIST. Systematic reporting of tumour risks and phenotypes in individuals with MINAS will facilitate the clinical interpretation of the significance of this diagnosis, which is becoming more frequent as strategies for genetic testing for hereditary cancer becomes more comprehensive.
Similar content being viewed by others


Introduction
Gastrointestinal stromal tumour (GIST) is a mesenchymal neoplasm arising in the gastrointestinal tract, most commonly occurring in the stomach or small intestine with around 10% in other locations [1]. DNA sequencing of GIST tissue reveals a somatic driver variant in KIT or PDGFRA in most cases but in 15% of adult cases and 85% of paediatric instances, no such driver is identified and the tumour is termed a wild-type GIST (wtGIST) [2, 3].
Amongst wtGISTs, more than three-quarters show evidence of succinate dehydrogenase (SDH) enzyme deficiency (known as dSDH-wtGIST), generally detectable by tumour immunostaining that reveals loss of SDHB expression [3, 4]. dSDH-wtGISTs show phenotypic differences to non-wtGIST including multinodular appearance, lymphovascular invasion, and epitheloid/mixed epitheloid spindle cell histology [5] and are less likely to respond to standard targeted therapy (e.g., imatinib) [2, 6]. They are frequently associated with a germline variants affecting the function of the SDHA, SDHB, SDHC or SDHD (SDHX genes) or somatic SDHC promoter hypermethylation. Whereas only 3–4% of non-wtGISTs occur in association with a causative germline variant (e.g. KIT, PDGFRA), one may be detected in as many as half of individuals with dSDH-wtGIST [3, 4, 7, 8]. The distribution of germline SDHX gene variants associated with dSDH-wtGIST differs from that seen in the tumour types most commonly associated with dSDH (phaeochromocytoma and paranglioma (PPGL) and head and neck paraganglioma) in that germline SDHA variants are frequent with wtGIST but an infrequent cause of PPGL or head and neck paraganglioma [7, 9]. Despite the high diagnostic yield for causative germline variants in individuals with wtGIST, reports of familial wtGIST are very rare. Whilst two sisters with dSDH-wtGIST and a germline variant affecting SDHA function have previously been reported [10], series of 34 (ref. [4]) and 33 (ref. [11]) wtGISTs reported no familial cases.
Here we describe a rare example of familial dSDH-wtGIST where a germline truncating SDHA variant was detected. In addition, a germline truncating PALB2 variant was also found meaning that two cancer predisposition gene variants with implications for genetic counselling were present in the same individual. This situation that has previously been termed multilocus inherited neoplasia alleles syndrome (MINAS). Whilst there is little to indicate that the PALB2 variant contributed to GIST tumourigenesis, we speculate that this unusual finding of may have led to increased penetrance in this family.
Materials (subjects) and methods
Participants
The family was ascertained via a clinical genetics centre after a mother and son presented with wtGIST. Information relating to these individuals has been submitted to the Leiden Open Variant Database and can be found at https://databases.lovd.nl/shared/diseases/04296 (individual IDs 00264003 and 00264004). Both affected individuals gave written informed consent for research investigations and the research was approved by the relevant Research Ethics Committees (South Birmingham REC and East of England—Cambridge South REC).
SDHB immunohistochemistry
SDHB immunohistochemistry was performed on 3-μm sections of FFPE tissue mounted on adhesive slides. The staining was performed on a fully automated BOND III IHC and ISH stainer system (Leica Biosystems, Nassloch, Germany). The SDHB primary antibody rabbit polyclonal (HPA002868, Sigma Aldrich, St Louis, MO, USA) was used. The Primary antibody binding to tissue sections was visualized using BOND Polymer Refine Detection system (DS9800, Leica Biosystems, Nassloch, Germany).
Whole-genome sequencing (WGS)
WGS and bioinformatic processing to produce variant call format (VCF) files was performed on samples from study participants as part of, and according to protocols devised by, the NIHR BioResource Rare Diseases study [12]. DNA Libraries were sequenced with an Illumina HiSeq 2500 instrument (Illumina Inc., San Diego, CA, USA). Read alignment to GRCh37 was performed using Illumina Isaac aligner version SAAC00776.15.01.27 [13]. Single nucleotide variants and indels were called from resulting binary compressed sequence alignment map (BAM) files using Illumina Starling software version 2.1.4.2.
Variant assessment from WGS data
Variants occurring in genes included in a list of 83 cancer predisposition genes were filtered and assessed according to a protocol described previously [14].
DNA extraction from formalin-fixed paraffin-embedded tumour blocks
Slides were prepared from formalin-fixed paraffin-embedded (FFPE) tumour blocks by the Human Research Tissue Bank, Cambridge University Hospitals. Following de-paraffinisation, slides were reviewed by a pathologist to mark selected tissue and tumour dissection was performed by colleagues in the Department of Haematology and Oncology diagnostic services, Cambridge University Hospitals. DNA was subsequently extracted from resulting tissue.
AmpliSeq panel sequencing
Library preparation was undertaken in the Stratified Medicine Core Laboratory using a custom AmpliSeq panel (Thermo Fisher Scientific, Waltham, MA, USA) that included the SDHA region of interest. An Illumina MiSeq instrument was used for sequencing. Alignment was performed with Burrows-Wheeler Aligner [15] and resulting BAM files were viewed with the Integrative Genomics Viewer [16].
Sanger sequencing
DNA extracted from tumours was also subject to Sanger sequencing for a PALB2 variant identified in the corresponding blood DNA according to standard protocols (see Supplementary material).
Variant description
Variants in this article are described based on transcripts NM_004168.4 for SDHA and NM_024675.3 for PALB2.
Results
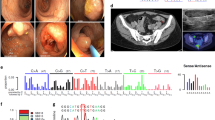
A 66-year-old female presented with symptoms of satiety and was endoscopically investigated, revealing a 35 mm gastric GIST. She went on to undergo a distal gastrectomy, at which time she was identified as having liver metastases. Further treatment was with imatinib. Subsequently, her son also developed a gastric GIST at the age of 34 years, which was detected as an incidental finding following imaging performed due to trauma. The 53 mm tumour was removed laparoscopically. Histology and immunohistochemistry of both GISTs showed a mixed epithelioid growth pattern and loss of SDHB staining on immunohistochemistry, indicating deficiency of a component of the succinate dehydrogenase (SDH) multiprotein complex (Fig. 1). There was no other reported family history of neoplasia and the proband’s other son had not been diagnosed with any tumours.

A, C Haematoxylin and eosin staining on samples from son and mother, respectively. B, D Loss of SDHB immunostaining on samples from son and mother, respectively. Non neoplastic cells show a retained mitochondrial staining pattern which can be appreciated in B and D.
The family was referred for Clinical Genetic assessment and initial routine analysis of SDHB, SDHC, SDHD, KIT and PDGFRA genes in the son’s sample did not reveal a causative constitutional variant and both individuals were recruited to research studies to try and elicit the cause. At the time of investigation (2014), analysis for SDHA variants or SDHC epimutations was not included in the diagnostic workflow. Following recruitment, the mother was also diagnosed with ductal carcinoma in situ of the breast at the age of 70 years.
WGS on DNA extracted from blood was performed on a research basis. Variant filtering based on a list of 83 cancer predisposition genes and variant assessment according to American College of Medical Genetics criteria revealed a nonsense variant in SDHA (c.1534C>T (p.Arg512*)) in both individuals. This variant has previously been reported as associated with GISTs and paragangliomas in the germline heterozygous state [11, 17] and has four submissions in ClinVar [18] with pathogenic or likely pathogenic assertion (two with paraganglioma as the condition for which the assertion is made, one with hereditary cancer predisposition, and one without a condition). Overall allele frequency in gnomAD [19] is 4.9 × 10−5 (seven occurrences) with a maximum allele frequency of 9.3 × 10−5 in non-Finnish Europeans.
Variant filtering and assessment also identified PALB2 c.3113 G>A (p.Trp1038*) in blood from both participants. PALB2 is a breast cancer predisposition gene and this is a frequently identified variant with >10 ClinVar entries with pathogenic assertion, mostly with reference to breast cancer predisposition. Overall allele frequency in gnomAD is 2.1 × 10−5 (two occurrences) with a maximum allele frequency of 3.1 × 10−5 in non-Finnish Europeans.
Loss of heterozygosity (LOH) analysis was performed on DNA from both tumours to support the role of the SDHA variant being causal and also investigate whether there was any evidence for the PALB2 variant contributing to tumourigenesis.
Loss of the SDHA wild-type allele was confirmed with a panel-based sequencing assay (see Materials (subjects) and methods section) where variant allele fraction (VAF) was 0.42 in the blood sample from the mother and 0.92 in her tumour sample. The son’s samples also showed some evidence of LOH with VAF’s of 0.57 in blood and 0.85 in tumour (Fig. 2). Loss of the wild-type PALB2 allele, which may have indicated a contribution to increased penetrance of the SDHA variant and occurrence in two family members, was not observed (Fig. 3) in either individual using Sanger sequencing

BAM files resulting from sequencing DNA extracted from tumour. Loss of SDHA wild-type allele shown in samples from both members of diad as indicated by predominance of variant (denoted by red colour) bases.

Retention of wild-type allele indicated by dual peak at site of variant.
Discussion
We report a rare case of familial dSDH-wtGIST that was associated with germline-predicted truncating variants in both SDHA and PALB2.
SDHA is a tumour suppressor gene and a second hit through loss of the wild-type allele can be observed in GISTs from germline variant carriers [3]. It encodes a catalytic subunit of the succinate dehydrogenase complex also comprised of components encoded by SDHB, SDHC and SDHD. The succinate dehydrogenase complex locates to the inner mitochondrial membrane and participates in the citric acid cycle by converting succinate to fumarate. Reduced function leads to accumulation of intracellular succinate and resultant inhibition of alpha-ketoglutarate-dependent dioxygenase enzymes including prolyl hydroxylase (PHD). PHD normally promotes hypoxia-inducible-factor 1-alpha (HIF1) breakdown through hydroxylation and reduction in this function leads to a pseudo-hypoxic state with associated upregulation of a range of genes that can promote neoplasia (e.g. IGF-1 and VEGF). Succinate accumulation also leads to oncogenic aberrant methylation via inhibition of TET enzymes [20] and has previously been observed in GISTs [21]. Alternative mechanisms to explain SDH deficiency contributing to tumourigenesis include altered amino acid metabolism and increased reactive oxygen species level as a result of mitochondrial dysfunction [5, 22].
Germline variants in SDHA, like other SDHX genes, can predispose to the development of PPGL but penetrance for these tumours is estimated to be under 10% from studies of relatives of probands diagnosed with PPGL and analysis of population databases [23,24,25]. Penetrance for GIST is not well defined but also appears to be low. Studies of 30 SDHA variant carriers ascertained by genetic testing for PPGL [23] and 95 carriers with PPGL identified in a literature review [24] each showed one GIST occurring in a family member of a case. Two GISTs were observed amongst ten SDHA germline variant carriers found through agnostic cancer genetic testing [26] and four GISTs occurred in 15 carriers identified through a review of UK genetic testing laboratory reports [27].
PALB2 encodes a protein that interacts with BRCA2 to execute double-stranded DNA repair through homologous recombination (HR) and was identified as a breast cancer predisposition gene in 2007 [28]. Penetrance by age 80 years for (female) breast cancer is currently estimated at 53% whilst the figures for ovarian and pancreatic cancers are 5% and 2–3%, respectively [29]. PALB2 is widely expressed at RNA and protein level, including in the gastrointestinal tract [30] but no association has previously been made with wtGIST.
Constitutional biallelic variants in PALB2 can cause Fanconi Anaemia associated with DNA repair deficiency. In individuals with monoallelic variants, a ‘second hit’ at the cellular level can lead to HR DNA repair deficiency and increased repair through the error-prone alternative mechanism of non-homologous end joining (NHEJ) [31]. These changes can be observed by analysis of tumour sequencing data including mutational signatures [32]. One signature is associated with biallelic inactivation of BRCA1 and BRCA2 (due to HR deficiency) but has also been demonstrated in breast [33] and pancreatic [34] cancers from individuals with constitutional PALB2 truncating variants. Around two-thirds of breast cancers from carriers of function compromising PALB2 variants have a second hit observed as either loss of the wild-type allele or a second somatic variant. Such biallelic deficits correlate closely with evidence of HR deficiency in tumours but tumours without an observable second hit can also occasionally have observable deficiency [31, 35], suggesting a second hit by an unidentified mechanism.
Given that familial wtGIST is rare, we performed further tumour studies to investigate whether there was evidence for the PALB2 variant contributing to increased penetrance as opposed to being an incidental finding. Tumour studies in both affected individuals supported the primary tumourigenic role of the SDHA variant with histology typical of SDH deficiency, loss of SDHB immunostaining (which indicates loss of a SDH complex component and is an indirect measure of SDHA loss) and evidence for loss of the wild-type SDHA allele on somatic sequencing. LOH analysis for PALB2 did not show evidence for loss of the wild-type allele. The PALB2 variant therefore, may have contributed to the breast cancer occurring in the mother (tumour tissue was unavailable for further analysis) but not the wtGISTs in this family. Preservation of the wild-type PALB2 allele in tumour studies supports that notion. However, absence of LOH in the context of a tumour suppressor gene does not always imply absent contribution as alternative mechanisms may disrupt the wild-type allele such as epimutation, structural variation not resulting in copy number loss, or a missense variant not assessed as contributory to tumourigenesis. More extensive tumour studies could potentially have been revealing in delineating the contribution of the PALB2 variant in these GISTs. IHC was useful to demonstrate the role of the SDHA variant but no equivalent assay was available for PALB2. Whole-genome sequencing was not performed due to inadequate DNA from FFPE tissue but may have revealed evidence of the HR deficient mutational signature that is characteristic of PALB2 related cancers. Interestingly, SDH deficiency has also been reported to suppress DNA repair by HR [36] so that signature could feasibly be observed in this scenario due to deficiency of either gene or enhanced by the presence of both.
To our knowledge, this is the first case of SDHA associated MINAS. The term describes the scenario in which an individual harbours clinically significant germline variants affecting more than one cancer predisposition gene and was suggested to facilitate information sharing about such cases [37]. This occurrence is often reported in the literature but the phenotypic effect is usually unclear, largely due to individual gene combinations being infrequently observed.
A key question following identification of a MINAS case is the nature of the resultant phenotype. Feasible effects include synergy between the variants to produce a more severe or penetrant phenotype, an independent manifestation where each variant leads to cancer risks equivalent to a scenario where the other is not present, and a protective effect due to synthetic lethality. We previously reviewed MINAS cases reported in the literature and generally observed evidence for an independent mechanism (including for the most frequent BRCA1/BRCA2 variant combination) but some cases show severe manifestations (early onset and/or atypical tumours) and cases with a protective effect are less likely to be investigated or reported.
Our studies suggest an independent action of the two variants where the PALB2 was an incidental finding though further studies would be required to confirm this. Regardless of any contribution to the wtGISTs, the situation produces challenges for genetic counselling in the sense that predictive testing and surveillance must be considered for two variants in the family. Individuals may have different perspectives regarding which of the variants to be tested for and there are likely to be uncertainties as to possible interactive effects between the variants that are difficult to delineate. These scenarios are becoming more frequent as broader genetic testing is applied in clinical settings and to alleviate these uncertainties through collation of cases, we encourage the reporting via the Leiden Open Variant Database (phenotypic tag MINAS) at https://databases.lovd.nl/shared/diseases/04296.
References
Rammohan A, Sathyanesan J, Rajendran K, Pitchaimuthu A, Perumal S-K, Srinivasan U, et al. A gist of gastrointestinal stromal tumors: a review. World J Gastrointest Oncol. 2013;5:102–12.
Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CDM, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Gr. J Clin Oncol. 2008;26:5360–7.
Boikos SA, Pappo AS, Killian JK, LaQuaglia MP, Weldon CB, George S, et al. Molecular Subtypes of KIT/PDGFRA Wild-Type gastrointestinal stromal tumors: a report from the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2016;2:922–8.
Janeway KA, Kim SY, Lodish M, Nosé V, Rustin P, Gaal J, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci USA. 2011;108:314–8.
Wang Y-M, Gu M-L, Ji F. Succinate dehydrogenase-deficient gastrointestinal stromal tumors. World J Gastroenterol. 2015;21:2303–14.
Ibrahim A, Chopra S. Succinate dehydrogenase-deficient gastrointestinal stromal tumors. Arch Pathol Lab Med. 2019;144:655–60.
Miettinen M, Lasota J. Succinate dehydrogenase deficient gastrointestinal stromal tumors (GISTs) - a review. Int J Biochem Cell Biol. 2014;53:514–9.
Casey RT, Ten Hoopen R, Ochoa E, Challis BG, Whitworth J, Smith PS, et al. SDHC epi-mutation testing in gastrointestinal stromal tumours and related tumours in clinical practice. Sci Rep. 2019;9:10244.
Bausch B, Schiavi F, Ni Y, Welander J, Patocs A, Ngeow J, et al. Clinical characterization of the pheochromocytoma and paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for gene-informed prevention. JAMA Oncol. 2017;3:1204–12.
Oudijk L, Gaal J, Korpershoek E, van Nederveen FH, Kelly L, Schiavon G, et al. SDHA mutations in adult and pediatric wild-type gastrointestinal stromal tumors. Mod Pathol. 2013;26:456–63.
Wagner AJ, Remillard SP, Zhang Y-X, Doyle LA, George S, Hornick JL. Loss of expression of SDHA predicts SDHA mutations in gastrointestinal stromal tumors. Mod Pathol. 2013;26:289–94.
Turro E, Astle WJ, Megy K, Gräf S, Greene D, Shamardina O, et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature. 2020;583:96–102.
Raczy C, Petrovski R, Saunders CT, Chorny I, Kruglyak S, Margulies EH, et al. Isaac: Ultra-fast whole-genome secondary analysis on Illumina sequencing platforms. Bioinformatics 2013;29:2041–3.
Whitworth J, Smith PS, Martin JE, West H, Luchetti A, Rodger F, et al. Comprehensive cancer-predisposition gene testing in an adult multiple primary tumor series shows a broad range of deleterious variants and atypical tumor phenotypes. Am J Hum Genet. 2018;103:3–18.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6.
Papathomas TG, Oudijk L, Persu A, Gill AJ, van Nederveen F, Tischler AS, et al. SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: a multicenter interobserver variation analysis using virtual microscopy: a Multinational Study of the European Network for the Study of Adrenal Tumors (ENS@T). Mod Pathol. 2015;28:807–21.
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–7.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Wang Q, Collins RL, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of- function intolerance across human protein-coding genes. BioRxiv. 2019.
Morin A, Goncalves J, Moog S, Castro-Vega L-J, Job S, Buffet A, et al. TET-mediated hypermethylation primes SDH-deficient cells for HIF2α-Driven mesenchymal transition. Cell Rep. 2020;30:4551–66.
Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Disco. 2013;3:648–57.
Dalla Pozza E, Dando I, Pacchiana R, Liboi E, Scupoli MT, Donadelli M, et al. Regulation of succinate dehydrogenase and role of succinate in cancer. Semin Cell Dev Biol. 2020;98:4–14.
van der Tuin K, Mensenkamp AR, Tops CMJ, Corssmit EPM, Dinjens WN, van de Horst-Schrivers ANA, et al. Clinical aspects of SDHA-related pheochromocytoma and paraganglioma: a nationwide study. J Clin Endocrinol Metab. 2018;103:438–45.
Maniam P, Zhou K, Lonergan M, Berg JN, Goudie DR, Newey PJ. Pathogenicity and Penetrance of Germline SDHA Variants in Pheochromocytoma and Paraganglioma (PPGL). J Endocr Soc. 2018;2:806–16.
Benn DE, Zhu Y, Andrews KA, Wilding M, Duncan EL, Dwight T, et al. Bayesian approach to determining penetrance of pathogenic SDH variants. J Med Genet. 2018;55:729–34.
Dubard Gault M, Mandelker D, DeLair D, Stewart CR, Kemel Y, Sheehan MR, et al. Germline SDHA mutations in children and adults with cancer. Cold Spring Harb Mol case Stud. 2018;4:a002584.
Casey RT, Ascher DB, Rattenberry E, Izatt L, Andrews KA, Simpson HL, et al. SDHA related tumorigenesis: a new case series and literature review for variant interpretation and pathogenicity. Mol Genet Genom Med. 2017;5:237–50.
Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–7.
Yang X, Leslie G, Doroszuk A, Schneider S, Allen J, Decker B, et al. Cancer risks associated with germline PALB2 pathogenic variants: an international study of 524 families. J Clin Oncol. 2019;38:674–85.
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419.
Li A, Geyer FC, Blecua P, Lee JY, Selenica P, Brown DN, et al. Homologous recombination DNA repair defects in PALB2-associated breast cancers. NPJ breast cancer. 2019;5:23.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio A, Behjati S, Biankin AV. et al. Signatures of mutational processes in human cancer. Nature . 2013;500:415–21.
Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet. 2017;49:1476–86.
Connor AA, Denroche RE, Jang GH, Timms L, Kalimuthu SN, Selander I, et al. Association of distinct mutational signatures with correlates of increased immune activity in pancreatic ductal adenocarcinoma. JAMA Oncol. 2017;3:774–83.
Lee JEA, Li N, Rowley SM, Cheasley D, Zethoven M, McInerny S, et al. Molecular analysis of PALB2-associated breast cancers. J Pathol. 2018;245:53–60.
Sulkowski PL, Sundaram RK, Oeck S, Corso CD, Liu Y, Noorbakhsh S, et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat Genet. 2018;50:1086–92.
Whitworth J, Skytte A-B, Sunde L, Lim DH, Arends MJ, Happerfield L, et al. Multilocus inherited neoplasia alleles syndrome: a case series and review. JAMA Oncol. 2015;2:373–9.
Acknowledgements
We thank NIHR BioResource volunteers for their participation, and gratefully acknowledge NIHR BioResource centres, NHS Trusts and staff for their contribution. We thank the National Institute for Health Research and NHS Blood and Transplant. This research was supported by the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care.
Funding
JW is supported by an NIHR Academic Clinical Lectureship. Funding for the National Institute for Health Research (NIHR) BioResource Rare Diseases project was provided by the NIHR (grant number RG65966). ERM acknowledges support from the European Research Council (Advanced Researcher Award), the NIHR (Senior Investigator Award and Cambridge NIHR Biomedical Research Centre), the Cancer Research UK Cambridge Cancer Centre, and the Medical Research Council (Infrastructure Award). The University of Cambridge has received salary support for ERM from the National Health Service (NHS) in the East of England through the Clinical Academic Reserve.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Members of the NIHR BioResource are listed in Supplementary information.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Whitworth, J., Casey, R.T., Smith, P.S. et al. Familial wild-type gastrointestinal stromal tumour in association with germline truncating variants in both SDHA and PALB2. Eur J Hum Genet 29, 1139–1145 (2021). https://doi.org/10.1038/s41431-021-00862-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00862-5
This article is cited by
-
Clinical genetics—it’s polygenic
European Journal of Human Genetics (2021)
